







专利摘要
本发明涉及大环脱氮-嘌呤酮衍生物、用于制备它们的方法、药物组合物、以及它们在治疗病毒感染中的用途。
权利要求
1.具有化学式(I)的化合物
及其药学上可接受的盐,其中
X是氧、氮或硫,
Y代表包含至少一个氮的芳族环或杂环,该芳族环或杂环任选地被独立地选自以下项的一个或多个取代基取代:C1-6烷基、C1-4烷氧基、三氟甲基或卤素,
Z代表任选地被烷基或烷基羟基取代的C1-10饱和或不饱和烷基;
或Z代表C1-6烷基-NH-C(O)-C1-6烷基-、C1-6烷基-NH-或C1-6烷基-NH-C(O)-C1-6烷基-O-;
或Z代表C1-10烷基-O-,其中所述烷基是不饱和的或饱和的并且可以任选地被烷基或烷基羟基取代,
或Z代表C1-6烷基-O-C1-6烷基-,其中所述烷基是饱和的或不饱和的并且可以任选地被烷基或烷基羟基取代
或Z代表C1-6烷基-O-C1-6烷基-O-,其中所述烷基是不饱和的或饱和的并且可以任选地被烷基或烷基羟基取代。
2.根据权利要求1所述的化合物,该化合物具有选自以下组中的以下化学式之一:
3.一种药物组合物,该药物组合物包括根据权利要求1或2所述的具有化学式(I)的化合物或其一种药学上可接受的盐、溶剂化物或多晶型物,连同一种或多种药学上可接受的赋形剂、稀释剂或载体。
4.根据权利要求1或2所述的具有化学式(I)的化合物或其一种药学上可接受的盐、溶剂化物或多晶型物,或根据权利要求3所述的药物组合物,用于用作一种药剂。
5.根据权利要求1或2所述的具有化学式(I)的化合物或其一种药学上可接受的盐、溶剂化物或多晶型物,或根据权利要求3所述的药物组合物,用于在治疗其中涉及TLR7调节的病症中使用。
说明书
本发明涉及大环脱氮-嘌呤酮衍生物、用于制备它们的方法、药物组合物、以及它们在治疗病毒感染中的用途。
本发明涉及大环脱氮-嘌呤酮衍生物在治疗病毒感染、免疫或炎性失调中的用途,由此涉及toll样受体(TLR)的调节或激动。Toll样受体是主要的跨膜蛋白质,其特征在于一种细胞外富含亮氨酸的结构域以及包含一个保守区域的细胞质延伸。先天免疫系统可以经由在某些类型的免疫细胞的细胞表面上表达的这些TLR来识别病原体相关分子模式。外来病原体的识别活化了细胞因子的产生以及在吞噬细胞上的共刺激分子的上调。这导致T细胞行为的调节。
估计到大部分哺乳动物种类具有十种与十五种之间的Toll样受体类型。已经在人类连同小鼠中鉴定了十三种TLR(即TLR1至TLR13),并且已经在其他哺乳动物物种中发现许多它们的等价型。然而,在人类中发现的某些TLR的等价型并不存在于所有哺乳动物中。例如,一种编码类似于人类中TLR10的蛋白质的基因存在于小鼠中,但是该基因显现出过去在某位点被一种反转录病毒损坏。另一方面,小鼠表达TLR11、TLR12以及TLR13,它们均不存在于人类中。其他的哺乳动物可以表达尚未在人类中发现的TLR。其他的非哺乳动物物种可以具有与哺乳动物不同的TLR,如TLR14(其在东方飩(Takifugupufferfish)中发现)所证实的。这可以使得使用实验动物作为人类先天免疫模型的方法复杂化。
对于toll样受体的详细综述参见以下杂志论文。霍夫曼,J.A(Hoffmann,J.A)《自然》(Nature)426,第33-38页,2003;阿基拉,S(Akira,S.),竹田,K(Takeda,K.)和贝少,T(Kaisho,T.)《免疫学年鉴》(AnnualRev.Immunology),21,第335-376页,2003;尤勒维奇,R.J.(Ulevitch,R.J.)《自然综述:免疫学》(NatureReviews:Immunology)4,第512-520页,2004。
先前已经描述了指示对Toll样受体有活性的化合物,如WO2006/117670中的嘌呤衍生物、WO98/01448和WO99/28321中的腺嘌呤衍生物、以及WO2009/067081中的嘧啶。
然而,存在一种强烈的对于新颖Toll样受体调节物的需要,这些调节物与现有技术的化合物相比具有优选的选择性、更高的效力、更高的新陈代谢稳定性以及改进的安全性。
在治疗某些病毒感染时,如同对于丙型肝炎病毒(HCV)的情况,可以给予干扰素(IFN-α)的定期注射。有关更多信息参见弗莱德(Fried)等人,用于慢性丙型肝炎病毒感染的聚乙二醇干扰素-α外加利巴韦林(Peginterferon-alfaplusribavirinforchronichepatitisCvirusinfection),《新英格兰医学杂志》(NEnglJMed)2002;347:975-82。可口服的小分子IFN诱导物提供了降低的免疫原性以及给予便利性的潜在优势。因此,新颖的IFN诱导物是用于治疗病毒感染的潜在有效的新类别药物。具有抗病毒效果的小分子IFN诱导物的一个实例,参见德克拉克,E.(DeClercq,E.);迪斯卡帕斯,J.(Descamps,J.);德索梅尔,P.(DeSomer,P.)《科学》(Science)1978,200,563-565。
在某些类型的癌症的治疗中还给出了与其他药物组合的IFN-α(《欧洲癌症杂志》(Eur.J.Cancer)46,2849-57,以及《癌症研究》(CancerRes.)1992,52,1056)。由于TLR7/8激动剂诱导显著的Th1应答的能力,它们也是感兴趣的疫苗佐剂(《人类疫苗》(Hum.Vaccines)2010,6,322-335,以及《人类疫苗》H(um.Vaccines)2009,5,381-394)。
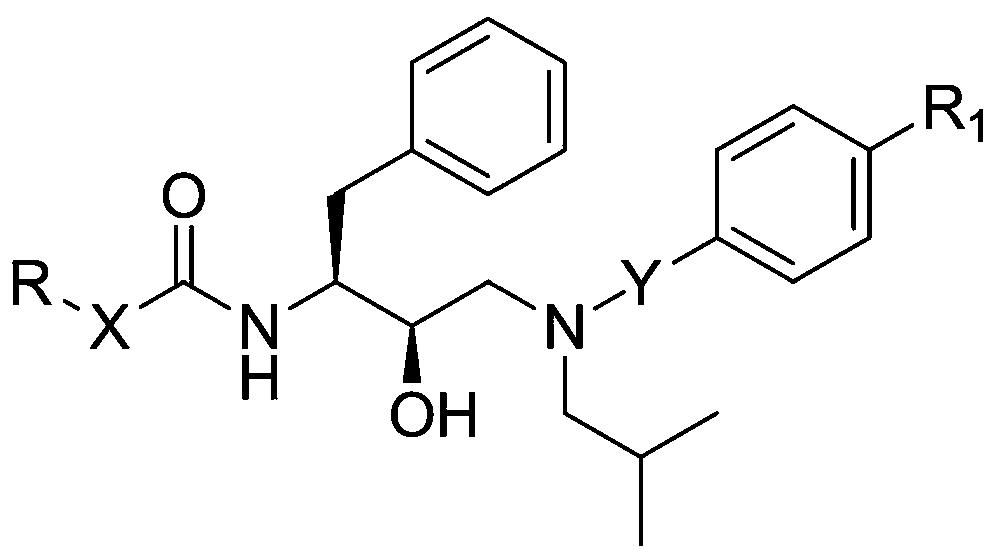
根据本发明提供了一种具有化学式(I)的化合物
及其药学上可接受的盐,其中
X是氧、氮或硫
Y表示包含至少一个氮的芳族环或杂环,该芳族环或杂环任选地被独立地选自以下的一个或多个取代基取代:C1-6烷基、C1-4烷氧基、三氟甲基或卤素,
Z代表任选地被烷基或烷基羟基取代的C1-10饱和或不饱和烷基;
或Z代表C1-6烷基-NH-C(O)-C1-6烷基-、C1-6烷基-NH-或C1-6烷基-NH-C(O)-C1-6烷基-O-;
或Z代表C1-10烷基-O-,其中所述烷基是不饱和的或饱和的并且可以任选地被烷基或烷基羟基取代,
或Z代表C1-6烷基-O-C1-6烷基-,其中所述烷基是饱和的或不饱和的并且可以任选地被烷基或烷基羟基取代
或Z代表C1-6烷基-O-C1-6烷基-O-,其中所述烷基是不饱和的或饱和的并且可以任选地被烷基或烷基羟基取代。
根据本发明具有以下化学式之一的优选化合物是选自下组:
本发明的一部分还是一种药物组合物,该药物组合物包含一种具有化学式(I)的化合物或其一种药学上可接受的盐、溶剂化物或多晶型物,连同一种或多种药学上可接受的赋形剂、稀释剂或载体。
另外属于本发明的是用作药剂的一种具有化学式(I)的化合物或其一种药学上可接受的盐、溶剂化物或多晶型物或者以上提到的一种药物组合物。
本发明还涉及用于在治疗一种其中涉及TLR7调节的病症中使用的一种具有化学式(I)的化合物或其一种药学上可接受的盐、溶剂化物或多晶型物或者以上提到的一种药物组合物。
术语“烷基”是指含有指定数目碳原子的直链或支链的大多数饱和的(但在根据本发明的特定化合物中是不饱和的)脂肪族烃。
术语“卤素”是指氟、氯、溴或碘。
术语“烷氧基”是指单独键合到氧的烷基(碳和氢链),例如像甲氧基基团或乙氧基基团。
具有化学式(I)的化合物的药学上可接受的盐包括其酸加成盐以及碱盐。合适的酸加成盐是从形成无毒盐的酸形成。合适的碱盐是从形成无毒盐的碱形成。
本发明的化合物还可以按非溶剂化的和溶剂化的形式存在。术语“溶剂化物”在此使用以描述包括本发明的化合物以及一种或多种药学上可接受的溶剂分子(例如,乙醇)的分子络合物。
术语“多晶型物”是指本发明的化合物以多于一种的形式或晶体结构存在的能力。
本发明的化合物可以按所谓的“互变异构体”形式存在,该互变异构体形式是指容易通过称为互变异构化的化学反应相互转化的有机化合物的异构体。这个反应导致氢原子或质子正式转移,伴随着单键和相邻双键的转变。
可以将本发明的化合物以晶态的或非晶态的产物给予。可以通过例如沉淀、结晶、冷冻干燥、喷雾干燥或蒸发干燥的方法获得呈固体塞、粉剂或膜的这些化合物。它们可以单独给予或与一种或多种本发明的其他化合物组合给予或与一种或多种其他药物组合给予。通常,它们将作为与一种或多种药学上可接受的赋形剂相结合的配制品而给予。术语“赋形剂”在此使用以描述除本发明的一种或多种化合物之外的任何成分。赋形剂的选择大体上取决于例如具体给予模式、赋形剂对溶解性和稳定性的影响以及剂型的性质等因素。
本发明的化合物或其任何亚群可制成各种给药目的药物制剂。作为合适的组合物可以引用所有通常用于全身给药的组合物。为制备本发明的药物组合物,将作为活性成分的有效量的具体化合物,任选为加成盐形式与药学上可接受的载体组合成紧密混合物,其载体根据给药需要的剂型可以采用各种形式。令人希望地,这些药物组合物处于适合于例如口服、经直肠或经皮给予的单位剂型。例如,在制备处于口服剂型的这些组合物中,可以采用任何常见药物介质,在口服液体制剂(例如悬浮液、糖浆剂、酏剂、乳液以及溶液)的情况下,诸如像,水、乙二醇、油、醇以及类似物;或在粉剂、丸剂、胶囊和片剂的情况下,固体载体,例如淀粉、糖、高岭土、稀释剂、润滑剂、粘合剂、崩解剂以及类似物。因为其容易给予,片剂和胶囊剂代表了最有利的口服单位剂型,在该情况下显然使用固体药物载体。还包括在使用之前不久可以被转变为液体形式的固体形式制品。在适用于经皮给药的组合物中,该载体可任选地包括穿透提髙剂和/或合适的润湿剂,任选地以较小比例结合任何性质的合适添加剂,所述添加剂不会对皮肤引入显著的有害作用。所述添加剂可促进向皮肤给药和/或可有助于制备期望的组合物。能以不同方式给予这些组合物,例如,作为透皮贴剂、作为点剂、作为软膏剂。还可以经由吸入或吹入法借助于在本域中采用的用于经由此方式给予的方法和配制品来给予本发明的化合物。因此,大体上,本发明的化合物能以溶液、悬浮液或干燥粉剂的形式而被给予至肺部。
为了便于给药和剂量的均一性,将上述药物组合物配制成单位剂型是特别有利的。如在此使用的单位剂型指的是适合作为单位剂量的物理离散单位,各单位含有预定量的活性成分,该预定量的活性成分经计算与所需药物载体相结合而产生所希望的治疗效果。此类单位剂型的实例是片剂(包括刻痕或包衣的片剂)、胶囊剂、丸剂、粉末包(powderpacket)、糯米纸剂(wafer)、栓剂、可注射溶液或悬浮液以及类似剂型,及其分离的多个。
在感染性疾病治疗领域中的普通技术人员将能够从下文所呈现的测试结果来确定有效量。通常认为有效日用量为从0.01mg/kg至50mg/kg体重,更优选从0.1mg/kg至10mg/kg体重。可适合地将所需剂量在1天中以合适的时间间隔分2个、3个、4个或更多个子剂量形式给予。所述子剂量可以被配制成单位剂型,例如每一单位剂型包含1至1000mg、且特别是5至200mg的活性成分。
如本领域的普通技术人员所熟知的,给予的精确剂量和频率取决于使用的具有化学式(I)的具体化合物、正在被治疗的具体病症、正在被治疗的病症的严重性、具体患者的年龄、体重和总体身体健康状况、连同个体可能服用的其他药物。此外,显然该有效量可以降低或增加,这取决于所治疗的受试者的反应和/或取决于对本发明的这些化合物开处方的医师的评估。因此以上所提及的有效量的范围仅仅是指导而不是旨在以任何程度限制本发明的范围或用途。
制备最终产物的总体方案:方法1
中间体B1的合成
在室温下,将SOCl2(80mL;1.11mol)逐滴添加到G1(19.1g;111mmol)在CH2Cl2(230mL)中的混合物里。将该混合物在室温下搅拌16h。蒸发溶剂,并且将残余物溶解于CH2Cl2中并用NaHCO3饱和水溶液处理直至碱性pH。将各层分离并用CH2Cl2萃取水层(两次)。将合并的有机层经MgSO4干燥,过滤并且在真空中浓缩以给出20.1g的棕色油。将该粗化合物不经进一步纯化而用于下一步骤中。
中间体C1的合成
将该反应在两个批次(13.6mmol和27.1mmol的A1)上平行地进行。
在室温下,向A1(5.0g;13.6mmol)和K2CO3(3.75g;27.1mmol)在丙酮(100mL)中的溶液里添加B1(4.46g;24.4mmol)和NaI(2.23g;14.9mmol)。将该混合物在室温下搅拌16h。将该混合物过滤并将滤液在真空中蒸发以给出一种棕色油。将两个批次合并并且通过制备型LC(不规则SiOH15-40μm,220g格瑞斯(Grace),流动相梯度:CH2Cl2/EtOAc从100/0至80/20)进行纯化。将含有产物的级分合并,并且将溶剂在真空中去除以给出18.6g的中间体C1(89%产率)。
中间体D1的合成
向C1(18.6g;36.1mmol)在THF(300mL)中的溶液里添加NH3水溶液(30%)(290mL),并且将该混合物在室温下搅拌16h。将该混合物用EtOAc和饱和NaCl溶液吸收,分离各层并且将有机层经MgSO4干燥,过滤并且将该溶剂在减压下去除以给出16.7g的黄橙色油。将该粗产物在高真空下干燥,以给出16.5g的粘性黄橙色的固体,将其直接用于下一步骤。
中间体E1的合成
在室温下,将NaH(60%在油中)(1.75g;43.7mmol)分部分地添加到烯丙醇(50mL)里。将该混合物在室温下搅拌30min之后,将其在0℃下逐滴添加到D1(5g;11.1mmol)在THF(124mL)中的溶液里。然后将所得混合物在室温下搅拌1h并且倾倒入饱和NH4Cl水溶液中。添加EtOAc和饱和NaCl水溶液,将各层进行分离并且将该水层用EtOAc萃取(一次)。合并的有机层经MgSO4干燥,过滤并且将溶剂在减压下去除,以给出一种黄色油。将该粗化合物通过制备型LC(不规则SiOH15-40μm,120g格瑞斯(Grace),液体注射,流动相梯度:从庚烷/EtOAc100/0至50/50)进行纯化,以给出4.04g的呈黄色油的中间体E1(79%产率)。
中间体F1的合成
以2个批次的850mg和2个批次的1g的E1进行该反应。
在此是用于一个批次的850mg的程序:
在一个舒伦克烧瓶中,将E1(0.85g;1.98mmol)和氯代二环己基硼烷(1M溶液在己烷中)(400μL;400μmol)在二氯乙烷(570mL)中的溶液在80℃在N2气氛下搅拌1h。添加格拉布-荷维达催化剂第2代(124mg;198μmol)并且将该混合物在120℃下搅拌16h。将该混合物通过N2鼓泡除气10min并且添加另外的格拉布-荷维达催化剂第2代(124mg;198μmol)和氯代二环己基硼烷(1M溶液在己烷中)(400μL;400μmol)。将该混合物在120℃下搅拌20h。
将2个批次混合并且添加钌清除剂(来自Silicycle的SiliaBondDMT)(10.4g;6.35mmol),并且将该混合物在室温下搅拌20h。将该反应混合物通过硅藻土垫过滤,并且将该溶剂在减压下去除,以给出一种棕色残余物。
将该残余物与从两个批次的1g的E1获得的残余物合并。将所得棕色残余物通过制备型LC(不规则SiOH15-40μm,120g格瑞斯(Grace),干燥装载,流动相梯度:从庚烷/EtOAc100/0至0/100)进行纯化,以给出1.19g的一种棕色固体。将该棕色固体通过制备型LC(固定相:不规则的裸二氧化硅40g,流动相梯度:从CH2Cl2/EtOAc90/10至80/20)进一步纯化,以给出705mg的一种黄色固体。将该黄色固体通过手性SFC(固定相:氨基6μm150x21.2mm,流动相:梯度从85%CO2、15%MeOH至65%CO2、35%MeOH)进一步纯化,以给出660mg的呈黄色固体的中间体F1(19%产率,E异构体)。
最终化合物1的合成
将F1(570mg;1.42mmol)和铁(795mg;14.2mmol)在AcOH(21mL)和水(4.2mL)中的混合物在50℃下搅拌2h。将该混合物浓缩至干燥。添加DMF,将该混合物进行超声处理、加热并通过硅藻土垫过滤,并且将该硅藻土用热DMF冲洗。将一种铁清除剂(来自SiliCycle的SiliaBond咪唑)(25.4g;29.5mmol)添加到该滤液中并且将该混合物在室温下搅拌16h。将该混合物通过硅藻土过滤,将该硅藻土用DMF冲洗并且将滤液在真空中进行浓缩,以给出620mg的棕色固体。将该粗品通过制备型LC(不规则SiOH,15-40μm,30g默克(Merck),流动相梯度:从CH2Cl2/MeOH/NH3aq98/2/0.2至85/15/1.5)进行纯化,以给出360mg的呈灰白色固体的最终化合物1(75%产率)。
中间体C1的替代合成
在0℃下,将偶氮二甲酸二异丙酯(DIAD)(3.0mL;15.0mmol)逐滴添加到A1(3.70g;10.028mmol)、G1(1.98g;12.0mmol)和PPh3(3.94g;15.0mmol)在THF(70mL)中的混合物里。将该混合物在室温下搅拌12h。添加EtOAc和水。倾析各层。将有机层用水洗涤,经MgSO4干燥,过滤并蒸发溶剂。将该粗品通过制备型LC在(不规则SiOH20-45μm450gMatrex)、流动相(85%庚烷,15%AcOEt)上进行纯化,以给出4.5g的中间体C1(87%产率)。
制备最终产物的总体方案:方法2
中间体H1的合成
将威尔金森催化剂(Wilkinson’scatalyst)(44mg;47.5μmol)添加到通过N2鼓泡吹扫15min的F1(190mg;475μmol)在THF/MeOH(50/50)(50mL)中的溶液里。将该混合物在室温下氢化(8巴)16h。将该混合物吹扫15min并进一步添加威尔金森催化剂(44mg;47.5μmol)。将该反应混合物在室温下氢化(8巴)4h。将该混合物在真空中进行浓缩,以给出一种棕色油。将该油通过制备型LC(不规则SiOH15-40μm,12g格瑞斯(Grace),干燥装载,流动相梯度:从CH2Cl2/EtOAc100/0至80/20)进行纯化,以给出150mg的呈黄色固体的中间体H1(79%产率)。
最终化合物2的合成
使用制备化合物1的程序来获得化合物2(54mg,44%产率)。
制备最终产物的总体方案:方法3
中间体J1的合成
将I1(5.9g;35.6mmol)添加到A1(7.3g;19.8mmol)、K2CO3(5.5g;39.6mmol)和NaI(3.3g;21.8mmol)在丙酮(145mL)中的溶液里。将该混合物在室温下搅拌20h。将该混合物通过硅藻土垫过滤并将滤液在真空中蒸发,以给出一种橙色固体。将该残余物吸收在CH2Cl2中。将沉淀过滤并且将滤液在真空中进行浓缩,以给出13g的一种黄色油。将该粗化合物通过制备型LC(不规则SiOH15-40μm,300gInterchim,流动相梯度:从庚烷/EtOAc100/0至80/20)进行纯化。将含有产物的级分进行合并,并且将溶剂在真空中去除,以给出7.1g的呈黄色油的中间体J1(72%产率)。
中间体K1的合成
在舒伦克烧瓶中,将J1(7.1g;14.2mmol)在THF(130mL)和NH3水溶液(30%)(130mL)中的溶液在室温下搅拌16h。将该混合物用EtOAc和NaCl饱和水溶液吸收,分离各层。将有机层经MgSO4干燥,过滤并且在真空中进行浓缩以给出6.4g的一种黄色油(100%产率)。
中间体L1的合成
在室温下和在N2气氛下,将NaH(2.2g;54.2mmol)分部分地添加到3-丁烯-1-醇(76mL)中。将该混合物在室温下搅拌30min之后,将其在0℃下逐滴添加到K1(5.9g;13.6mmol)在THF(150mL)中的溶液里。将所得混合物在0℃下搅拌1h。将混合物倾倒入饱和NH4Cl水溶液中。添加EtOAc和饱和水性NaCl溶液,将各层进行分离。将有机层经MgSO4干燥,过滤并且在真空中进行浓缩以给出一种黄色残余物,将该残余物与甲苯共沸蒸馏(一次),以给出6.6g的一种黄色油。将该粗化合物通过制备型LC(不规则SiOH15-40μm,220g格瑞斯(Grace),流动相梯度:从庚烷/EtOAc100/0至50/50)进行纯化。将含有产物的级分进行合并,并且将溶剂在真空中去除,以给出4.46g的呈黄色油的中间体L1(77%产率)。
中间体M1的合成
将该反应以2个批次进行。
对于一个批次的典型程序如下:
将L1(2.45g;5.75mmol)在无水CH2Cl2(1.7L)中的溶液通过N2鼓泡除气15min。添加格拉布催化剂第2代(488mg;574μmol)并且将该混合物在室温下搅拌72h。添加SiliaBondDMT(7.66g;4.59mmol)并且将该混合物在室温下搅拌16h。将2个批次合并并且通过硅藻土过滤。将该滤液在真空中进行浓缩,以给出一种黑色固体。将该粗化合物通过制备型LC(不规则SiOH15-40μm,150g默克(Merck),流动相梯度:从庚烷/EtOAc100/0至50/50)进行纯化。将含有产物的级分进行合并,并且将溶剂在真空中去除,以给出230mg的级分1和2.3g的级分2。将级分2通过制备型LC(固定相:不规则SiOH40μm120g,流动相:庚烷/CH2Cl2/MeOH55/43/2)进行纯化。将分离的化合物与级分1合并并且通过手性SFC(固定相:ChiralpakIC5μm250x20mm,流动相:70%CO2,30%iPrOH)进行纯化,以给出1.51g的呈黄色固体的中间体M1(33%产率,异构体E)。
最终化合物3的合成
将铁(631mg,11.3mmol)添加到D4(750mg;1.88mmol)在AcOH(150mL)和水(25mL)中的溶液里。将该混合物在80℃下搅拌16h。添加铁(315mg,5.65mmol)并且将该混合物在80℃下搅拌2h。添加铁(315mg,5.65mmol)并且将该混合物在80℃下搅拌4h。添加铁(315mg,5.65mmol)并且将该混合物在80℃下搅拌16h。将该混合物浓缩至干燥。添加DMF,将该混合物通过硅藻土过滤,并且将该硅藻土用热DMF冲洗。将SiliaBond咪唑(48.7g;56.5mmol)添加到该滤液中并且将该混合物在室温下搅拌16h。将该混合物通过硅藻土过滤,将该硅藻土用DMF冲洗并且将滤液在真空中进行浓缩。将该粗化合物通过制备型LC(不规则SiOH,15-40μm,25g默克(Merek),流动相梯度:从CH2Cl2/MeOH/NH3aq98/2/0.2至85/15/1.5)进行纯化,以给出2个级分。将级分1用EtOH吸收并过滤以给出级分3,并且将级分2用MeCN吸收并过滤以给出级分4。将级分3和级分4合并在EtOH中,过滤并在真空中干燥,以给出199mg的最终化合物3(33%产率)。
制备最终产物的总体方案:方法4
中间体N1的合成
将威尔金森催化剂(46mg;50.2μmol)添加到通过N2鼓泡吹扫15min的M1(200mg;502μmol)在THF/MeOH(50/50)(50mL)中的溶液里。将该混合物在室温下氢化(7巴)20h。将该混合物通过N鼓泡吹扫15min,添加另外的威尔金森催化剂(46mg;50.2μmol),并且将该反应混合物在室温下氢化(7巴)16h。将该反应混合物在真空中进行浓缩,以给出一种绿色油。将该油通过制备型LC(不规则SiOH15-40μm,25g默克(Merck),干燥装载,流动相梯度:从庚烷/EtOAc100/0至70/30)进行纯化,以给出130mg的呈黄色固体的中间体N1(66%产率)。
中间体O1的合成
在压力容器反应器中,将N1(110mg;275μmol)在EtOH(5mL)中用Pd/C(10%)(30mg;28.5μmol)作为催化剂在40℃下氢化(3巴)6h。将催化剂通过经硅藻土过滤去除,将硅藻土用EtOH洗涤并且将滤液在真空下蒸发,以给出100mg的一种黄色残余物(98%产率)。将中间体O1不经进一步纯化而用于下一步骤中。
最终化合物4的合成
在密封管中,将在纯乙酸(5mL)中的O1(100mg;270μmol)在室温下搅拌90min。将溶剂在减压下除去,以给出一种黄色残余物。将残余物用CH2Cl2吸收并且将该溶剂在减压下去除(两次),以给出87mg的一种黄绿色固体。将该固体与甲苯共沸蒸馏(四次),并且然后在Et2O中进行研磨和超声处理。将该混合物过滤出(玻璃料n°5),以给出75mg的最终化合物4(77%产率,乙酸盐)。
制备最终产物的总体方案:方法5
中间体Q1的合成
在室温下,向A1(3.52g;9.54mmol)和K2CO3(2.64g;19.1mmol)在丙酮(80mL)中的溶液里添加P1(1.93g;10.5mmol)和NaI(1.57g;10.5mmol)。将该混合物在室温下搅拌16h,添加另外的P1(1.5g;8.17mmol)并且将该混合物在室温下搅拌24h。将该反应混合物通过硅藻土垫过滤并将滤液在真空中蒸发,以给出一种黑色残余物。将该残余物通过制备型LC(不规则SiOH15-40μm,80g格瑞斯(Grace),干燥装载,流动相梯度:从庚烷/EtOAc100/0至50/50)进行纯化,以给出3.28g的呈橙色油的中间体Q1(67%产率)。
中间体R1的合成
在舒伦克烧瓶中,在室温下向Q1(3.28g;6.36mmol)在THF(52mL)中的溶液里添加NH3水溶液(30%)(52mL)。将该混合物在室温下搅拌26h,并且添加另外的NH3水溶液(10mL)并且将该混合物在室温下搅拌4h。将该混合物用EtOAc和NaCl饱和水溶液吸收,分离各层并且将有机层经MgSO4干燥,过滤并且将该溶剂在减压下去除以给出2.74g的呈黄色油的中间体R1(87%产率)。
中间体S1的合成
在室温下,将NaH(60%在油中)(888mg;22.2mmol)分部分地添加到3-丁烯-1-醇(30mL;354mmol)中。将该混合物在室温下搅拌30min之后,将其在0℃下逐滴添加到R1(2.74g;5.63mmol)在THF(62mL)中的溶液里。将所得混合物在室温下搅拌1h并且将其倾倒入NH4Cl饱和水溶液中。添加EtOAc和NaCl饱和水溶液,将各层进行分离并且将水层用EtOAc(一次)萃取。合并的有机层经MgSO4干燥,过滤并且将溶剂在减压下去除,以给出一种黄色油。将该油通过制备型LC(不规则SiOH15-40μm,80g格瑞斯(Grace),干燥装载,流动相梯度:从庚烷/EtOAc100/0至20/80)进行纯化,以给出1.06g的呈黄色残余物的中间体S1(42%产率)。
中间体T1的合成
将该反应以两个批次的480mg的中间体S1进行。
在此报道用于一个批次的程序:
在舒伦克烧瓶中,将S1(480mg;1.08mmol)和氯代二环己基硼烷(1M在己烷中)(216μL;216μmol)在无水二氯乙烷(300mL)中的溶液在80℃下和在N2气氛下搅拌1h。添加格拉布-荷维达催化剂第2代(68mg;108μmol)并且将该混合物在120℃下搅拌2h。
将两个批次混合,添加SiliaBondDMT(2.84g;1.73mmol)并且将该混合物在室温下搅拌20h。
将该混合物通过硅藻土垫过滤,将该硅藻土用EtOAc洗涤并且将滤液在真空中蒸发,以给出一种棕色固体。将该棕色固体通过制备型LC(不规则SiOH15-40μm,40g格瑞斯(Grace),干燥装载,流动相梯度:CH2Cl2/EtOAc从100/0至20/80)进行纯化,以给出610mg的一种黄色残余物(E和Z异构体的混合物,中间体U1)。将310mg的中间体U1通过反相(固定相:Nucleodur-Sphinxrp5μm21x150mm,流动相:梯度从70%甲酸0.1%、30%MeCN至0%甲酸0.1%、100%MeCN)进行纯化,以给出195mg的呈黄色固体的中间体T1(E异构体)(22%产率)。
最终化合物5的合成
将T1(160mg;385μmol)和铁(129mg;2.31mmol)在乙酸(21mL)和水(2.4mL)中的混合物在80℃下搅拌7h。添加另外的铁(129mg,2.31mmol)并且将该混合物在80℃下搅拌16h。添加另外的铁(129mg,2.31mmol)并且将该混合物在80℃下搅拌3h。将该混合物在真空中进行浓缩,以给出一种残余物。将该残余物稀释于DMF中并通过硅藻土垫过滤。将SiliaBond咪唑(12.7g;14.7mmol)添加到该滤液中并且将该混合物在室温下搅拌16h。将该混合物通过硅藻土垫过滤并将滤液在真空中蒸发,以给出一种棕色固体。将该棕色固体通过制备型LC(不规则SiOH15-40μm,12g格瑞斯(Grace),干燥装载,流动相梯度:从CH2Cl2/MeOH/NH3aq97/3/0.3至80/20/2)进行纯化,以给出65mg的灰白色固体。将该固体通过反相(固定相:X-桥-C185μm30*150mm,流动相梯度:从H2O(0.5%NH4CO3)/MeCN70/30至0/100)进行纯化,以给出43mg的呈白色固体的最终化合物5(31%产率,E异构体)。
制备最终产物的总体方案:方法6
中间体V1的合成
将威尔金森催化剂(58mg;62.6μmol)添加到通过N2鼓泡吹扫15min的U1(Z/E混合物,260mg;626μmol)在THF/MeOH(50/50)(66mL)中的溶液里。将该混合物在室温下氢化(7巴)16h。添加另外的威尔金森催化剂(58mg;62.6μmol)并且将该混合物在室温下氢化6h。将该反应混合物在真空中进行浓缩,以给出一种棕色固体。将该固体通过制备型LC(不规则SiOH15-40μm,25g默克(Merck),干燥装载,流动相梯度:从庚烷/EtOAc100/0至50/50)进行纯化,以给出250mg的呈黄色油的中间体V1(54%产率)。
最终化合物6的合成
将V1(238mg;359μmol)和铁(120mg;2.16mmol)在乙酸(20mL)和水(2.2mL)中的混合物在80℃下搅拌6h。添加另外的铁(120mg,2.16mmol)并且将该混合物在80℃下搅拌20h。添加另外的铁(120mg,2.16mmol)并且将该混合物在80℃下搅拌5h。将该混合物在真空中进行浓缩,以给出一种残余物。将该残余物稀释于DMF中并通过硅藻土垫过滤。将SiliaBond咪唑(11.1g;12.9mmol)添加到该滤液中并且将该混合物在室温下搅拌16h。将该混合物通过硅藻土垫过滤并将滤液在真空中蒸发,以给出一种棕色固体。将该固体通过制备型LC(不规则SiOH15-40μm,12g格瑞斯(Grace),干燥装载,流动相梯度:从CH2Cl2/MeOH/NH3aq97/3/0.3至80/20/2)进行纯化,以给出32mg的灰白色固体。将该固体用水吸收,进行研磨和超声处理。将所得悬浮液过滤出(玻璃料n°5)中并且用Et2O洗涤(两次),以给出19mg的呈灰白色固体的最终化合物6(15%产率)。
制备最终产物的总体方案:方法7
中间体W1的合成
在室温下,将NaH(60%在油中)(2.1g;52.1mmol)分部分地添加到3-丁烯-1-醇(74mL)中。将该混合物在室温下搅拌30min之后,将其在0℃下逐滴添加到D1(5.97g;13.2mmol)在THF(150mL)中的溶液里。将所得混合物在室温下搅拌2h30min并且倾倒入NH4Cl水性饱和溶液中。添加EtOAc和NaCl饱和水溶液,将各层进行分离并且将该水层用EtOAc萃取(一次)。将合并的有机层经MgSO4干燥,过滤并在减压下除去溶剂,以给出6.77g的一种黄色油。将该粗品通过制备型LC(不规则SiOH15-40μm,120g格瑞斯(Grace),液体注射,流动相梯度:从庚烷/EtOAc100/0至50/50)进行纯化,以给出5.12g的呈黄色油的中间体W1(83%产率)。
中间体X1的合成
在室温下,向通过N2鼓泡除气15min的W1(3g;6.78mmol)在CH2Cl2(1.3L)中的溶液里添加格拉布催化剂第2代(578mg;678μmol)。将该溶液在室温下搅拌20h。添加SiliaBondDMT(8.89g;5.42mmol)并且将该混合物在室温下搅拌20h。将该反应混合物通过硅藻土垫过滤并且将溶剂在减压下去除,以给出一种褐色残余物,将其与另一批合并(0.226mmol规模)。将合并的残余物用MeOH吸收,进行超声处理并加热,以给出一种沉淀,将其过滤出以给出3.2g的一种褐色固体。将该粗品通过制备型LC(不规则SiOH,15-40μm,220g格瑞斯(Grace),干燥装载,流动相梯度:从CH2Cl2/EtOAc100/0至50/50)进行纯化,以给出1.7g的呈淡棕色固体的级分1。将级分1用MeOH吸收,进行超声处理并加热,以给出一种沉淀,将其过滤出以给出820mg的呈淡棕色固体的级分2。
将滤液在真空中进行浓缩,以给出590mg的呈棕色残余物的级分3(不纯的X1)。将级分2通过制备型LC(固定相:球形裸二氧化硅5μm,150x30.0mm,流动相梯度:从庚烷/EtOAc85/15至0/100)进行纯化,以给出435mg的呈黄色固体的中间体X1(E异构体,15%产率)。
将级分3与另一批次进行纯化。
最终化合物10的合成
将V1(430mg;1.04mmol)和铁(579mg;10.4mmol)在乙酸(43mL)和水(3mL)中的混合物在50℃下搅拌4h。将该混合物浓缩至干燥。添加DMF。将该混合物进行超声处理,加热并且通过硅藻土垫过滤并且该硅藻土用热DMF冲洗。将SiliaBond咪唑(17.9g;20.8mmol)添加到该滤液中并且将该混合物在室温下搅拌16h。将该混合物通过硅藻土过滤,将该硅藻土用DMF冲洗并且将滤液在真空中进行浓缩,以给出670mg的粗化合物。将该粗品通过制备型LC(不规则SiOH,15-40μm,25g默克(Merck),流动相梯度:从CH2Cl2/MeOH/NH3aq98/2/0.2至85/15/1.5)进行纯化,以给出一种灰白色固体。在20h期间,将该固体在40℃在减压下干燥,以给出295mg的呈灰白色固体的最终化合物10(84%产率)。
制备最终产物的总体方案:方法8
中间体Y1的合成
将威尔金森催化剂(103mg;111μmol)添加到通过N2鼓泡吹扫15min的X1(230mg;0.555mmol)在THF/MeOH(50/50)(40mL)中的溶液里。将该混合物在室温下氢化(8巴)24h。将该反应混合物在真空中进行浓缩,以给出一种棕色残余物。将该固体通过制备型LC(不规则SiOH,15-40μm,12g格瑞斯(Grace),干燥装载,流动相梯度:从CH2Cl2/EtOAc100/0至90/10)进行纯化,以给出55mg的呈黄色残余物的中间体Y1(24%产率)。
最终化合物14的合成
将Y1(55mg;0.132mmol)和铁(74mg;1.32mmol)在乙酸(5.5mL)和水(0.4mL)中的混合物在50℃下搅拌20h。添加更多的铁(37mg,0.66mmol)并且将该混合物在50℃下搅拌3h。添加更多的铁(37mg,0.66mmol)并且将该混合物在50℃下搅拌20h。将该混合物通过硅藻土垫过滤并且该硅藻土用乙酸冲洗。将更多的铁(74mg;1.32mmol)添加到该滤液中并且将该混合物在50℃下搅拌88h。将更多的铁(74mg;1.32mmol)添加到该滤液中并且将该混合物在80℃下搅拌24h。该环化作用未完成。将该混合物在真空中进行浓缩,以给出一种棕色固体。
将TiCl3(8.60mL;10.0mmol)逐滴添加到该棕色固体在C(19mL)中的溶液里。将该混合物在室温下搅拌过夜。在0℃下,将该混合物通过添加K2CO3粉末进行碱化。将所得混合物通过硅藻土垫过滤,并且将该硅藻土用AcOEt/MeOH(8∶2)的溶液洗涤。将滤液真空进行浓缩。将该粗固体通过制备型LC(不规则SiOH,15-40μm,10g默克(Merck),干燥装载,流动相梯度:从CH2Cl2/MeOH/NH3aq98/2/0.2至85/15/1.5)进行纯化。将含有产物的级分进行合并,并且将溶剂在真空中去除,以给出20mg的呈灰白色固体的最终化合物14(12%产率)。
制备最终产物的总体方案:方法9
中间体A2的合成
将甲磺酰氯(8.4mL;108mmol)添加到Z1(14g;72.1mmol)、NEt3(20mL;144mmol)和LiCl(4.6g;108mmol)在无水CH2Cl2(980mL)中的溶液里。将该混合物在室温下搅拌1h30。添加水并且将各层进行分离。将有机层用水洗涤(一次),经MgSO4干燥,过滤并且在真空中进行浓缩,以给出18.8g的呈绿色油的A2(96%)。
中间体B2的合成
使用针对中间体C1所描述的程序来获得中间体B2(78%产率,呈一种黄色油)。
中间体C2的合成
使用针对中间体D1所描述的程序来获得中间体C2(定量产量,呈一种黄色油)。
中间体D2的合成
使用针对中间体W1所描述的程序来获得中间体D2(64%产率,呈一种黄色固体)。
中间体E2的合成
将D2(1g;2.12mmol)在CH2Cl2(400mL)中的溶液通过N2鼓泡除气15min。添加格拉布催化剂第2代(181mg;212μmol)并且将该混合物在室温下搅拌16h。添加SiliaBondDMT(2.78g;1.69mmol)并且将该混合物在室温下搅拌16h。将该混合物通过硅藻土垫过滤并将滤液在真空中进行浓缩,以给出1.11g的一种棕色油。将该粗品通过制备型LC(不规则SiOH15-40μm,50g默克(Merck),流动相梯度:从CH2Cl2/EtOAc100/0至90/10)进行纯化。将含有产物的级分进行合并,并且将溶剂在真空中去除,以给出386mg的呈黄色泡沫的中间体E2(41%,异构体E(96.2%)+异构体Z(3.8%))。
最终化合物15的合成
将铁(291mg;5.21mmol)添加到E2(386mg;0.869mmol)在乙酸(36mL)和水(3mL)中的溶液里。将该混合物在80℃下搅拌6h。添加铁(146mg,2.61mmol)并且将该混合物在80℃下搅拌16h。再次添加铁(146mg,2.61mmol)并且将该混合物在80℃下搅拌5h。将该混合物浓缩至干燥。添加DMF,将该混合物通过硅藻土过滤,并且将该硅藻土用DMF冲洗。将Siliabond咪唑(18g;20.9mmol)添加到该滤液中并且将该混合物在室温下搅拌72h。将该混合物通过硅藻土过滤,将该硅藻土用DMF冲洗并且将滤液在真空中进行浓缩,以给出428mg的棕色固体。将该残余物吸收在CH3CN中,从而导致沉淀。将该沉淀过滤以给出267mg的一种棕色固体。将该固体通过制备型LC(不规则SiOH15-40μm,10g默克(Merck),干燥装载,流动相梯度:从CH2Cl2/MeOH/NH3aq95/5/0.5至85/15/1.5)进行纯化。将含有产物的级分进行合并,并且将溶剂在真空中去除,以给出124mg的一种灰白色固体。将该固体通过反向(固定相:Sunfire-C185μm19x150mm,流动相梯度:从CH3CN/H2O(甲酸0.1%)5/95至50/50)进行纯化,以给出72mg的呈白色固体的最终化合物15(23%产率)。
制备最终产物的总体方案:方法10
中间体F2的合成
使用针对中间体F1(E异构体)所描述的程序来获得中间体F2。
最终化合物16的合成
在室温下,将TiCl3(12.3mL;14.341mmol)逐滴添加到F2(300mg;0.717mmol)在THF(30mL)中的混合物里。将该混合物在室温下搅拌2小时。将该混合物冷却至0℃并且用K2CO3粉末进行碱化。将所得泥质混合物通过硅藻土垫过滤,并且将该硅藻土用AcOEt/CH3OH8/2的溶液洗涤。将滤液部分蒸发,以便在过滤白色固体并在真空压力下在85℃干燥之后给出175mg的最终化合物16(71%产率)。
最终化合物17的合成
将该盐酸盐用10当量的HCl(4N在二噁烷中)来制备,将其添加到化合物16(100mg;0.292mmol)在CH3OH中的悬浮液里(10mL)。将沉淀物搅拌3h,过滤并在真空下在90℃干燥过夜。将该固体溶解在MeOH/CH2Cl250/50中,添加CH3CN并且将该溶剂进行蒸发直到一种白色固体沉淀,将其过滤并且在真空下在90℃干燥,以给出47mg的呈HCl盐的最终化合物17(0.93HCl,0.51H2O;42%产率)。
制备最终产物的总体方案:方法11
中间体H2的合成
在-20℃下在N2流下,将在THF(100mL)中的G2(22.0g;72.04mmol)逐滴添加到LiAlH4(3.28g;86.45mmol)在THF(120mL)中的悬浮液中。将该混合物在0℃下搅拌1h。逐滴添加3.5mL的水,随后添加3.5mL的NaOH3N和10mL的水。将所得混合物通过硅藻土垫过滤并且该硅藻土用EtOAc洗涤。将滤液在减压下进行浓缩,以给出19g的呈黄色油的中间体H2(定量产量)。
中间体I2的合成
在0℃下,将偶氮二甲酸二异丙酯(4.0mL;20.32mmol)逐滴添加到A1(5.0g;13.55mmol)、H2(4.28g;16.26mmol)和PPh3(5.33g;20.327mmol)在THF(100mL)中的混合物里。将该混合物在室温下搅拌12h。添加EtOAc和水。倾析各层。将有机层用水洗涤,经MgSO4干燥,过滤并蒸发溶剂。将该粗化合物溶解在庚烷/EtOAc80/20中,将沉淀物过滤出(主要是POPh3)并且将滤液通过层析法进行纯化。通过快速层析法在硅胶(15-40μm,220g,庚烷/EtOAc80/20)上进行纯化。将纯的级分进行收集并蒸发至干燥,以给出8.2g中间体I2(99%产率)。
中间体J2的合成
在室温下,将I2(8.2g,13.349mmol)在NH4OH(100mL)和THF(100mL)中搅拌24h。将该混合物在减压下进行半蒸发。将残余物用EtOAc吸收。将有机层用水洗涤,经MgSO4干燥,过滤并且蒸发溶剂,以给出8.15g的中间体J2(定量产量)。将该粗化合物直接用于下一个反应步骤中。
中间体K2的合成
在N2流下,在室温下,将NaH(60%在油中)(1.15g;28.64mmol)分部分地添加到烯丙醇(35mL)中。将该混合物在室温下搅拌30min之后,将其在0℃下逐滴添加到J2(4.0g;7.26mmol)在THF(80mL)中的溶液里。然后将所得混合物在室温下搅拌2h30min并且倾倒入NH4Cl饱和溶液中。添加EtOAc和NaCl饱和水溶液,将各层进行分离并且将该水层用EtOAc萃取(一次)。将合并的有机层经MgSO4干燥,过滤并在减压下除去溶剂,以给出4.7g的一种黄色油。通过快速层析法在硅胶(15-40μm,80g,CH2Cl2/庚烷65/35)上进行纯化。将纯的级分进行收集并蒸发至干燥,以给出2.65g中间体K2(69%产率)。
中间体L2的合成
在反应之前,将二氯乙烷通过鼓泡N2通过来脱气。
在Slenck管中,将K2(1.3g;2.464mmol)和氯代二环己基硼烷(1M在己烷中)(493μL;0.493mmol)在二氯乙烷(600mL)中的溶液在80℃在N2下搅拌1h。添加格拉布-荷维达催化剂第2代(609mg;0.493mmol)并且将该混合物在120℃下搅拌16h。添加SiliabondDMT(2.98g;1.82mmol)并且将该混合物在室温下搅拌16h。将该反应混合物通过硅藻土过滤并且将滤液在真空中蒸发,以给出1.6g,将其与另一反应(2.46mmol规模)进行合并,之后进行纯化(纯化3.2g的总重量)。通过快速层析法在硅胶(15-40μm,80g,CH2Cl2/CH3OH99.5/0.5)上进行纯化。将纯的级分进行收集并蒸发至干燥,以给出0.99g的F1(预期化合物的E/Z混合物,40%产率)和0.65g的F2(起始材料K2)。
将F1通过手性SFC(固定相:NH25μm150*30mm,流动相:92%CO2,8%MeOH)进一步纯化,以给出664mg的中间体L2(E异构体,27%产率)。
中间体M2的合成
将铁(1.45g;26.025mmol)添加到L2(0.65g;1.301mmol)在乙酸(15mL)和水(1.5mL)中的混合物里。将该混合物在50℃下搅拌3h,并且然后用CH2Cl2/MeOH通过硅藻土过滤。将滤液在减压下进行浓缩。将该化合物通过快速硅胶柱层析(15-40μm;80g,洗脱液CH2Cl2/CH3OH/NH4OH96/4/0.5)进行纯化,以给出640mg。通过快速层析法在硅胶(15-40μm,40g,CH2Cl2/CH3OH/NH4OH:97/3/0.2)上进行第二次纯化。将纯的级分进行收集并蒸发至干燥,以给出240mg中间体M2(38%产率)。
最终化合物18的合成
在0℃下,将CF3CO2H(0.455mL)逐滴添加到M2(100mg,0.236mmol)在CH2Cl2(1mL)中的混合物里。将该混合物在室温下搅拌过夜,并且然后用K2CO3在水中的10%溶液进行碱化。将沉淀物过滤出,用水和CH3CN洗涤,并且最终在真空下干燥,以提供35mg的最终化合物18(E异构体,46%产率)。
制备最终产物的总体方案:方法12
中间体N2的合成
将M2(140mg,0.331mmol)在THF/CH3OH(50/50)(30mL)中的混合物在10巴压力下用威尔金森催化剂(61.2mg,0.0661mmol)氢化72h。添加SiliabondDMT(441mg,0.264mmol)并且将该混合物在室温下搅拌18h。将该混合物通过硅藻土垫过滤,并且将该硅藻土用CH2Cl2/CH3OH95/5洗涤。将滤液在减压下进行浓缩。通过快速层析法在硅胶(15-40μm,10g,CH2Cl2/CH3OH/NH4OH:97/3/0.1)上进行纯化。将纯的级分进行收集并蒸发至干燥,以给出62mg中间体N2(44%产率),将其按原样用在下一步骤中。
最终化合物23的合成
在0℃下,将CF3CO2H(0.281mL,3.643mmol)逐滴添加到N2(62mg,0.146mmol)在CH2Cl2(1mL)中的混合物里。将该混合物在室温下搅拌过夜。将该混合物用K2CO3在水中的10%溶液进行碱化。将该混合物用CH2Cl2和CH3OH(80/20)萃取两次。将有机层经MgSO4干燥,过滤,并蒸发溶剂。将该粗化合物用DMF吸收,添加2g的SiO260-200μm并且将所得悬浮液蒸发至干燥。将这残余物放在层析柱的顶部(固体沉积物)。通过快速层析法在硅胶(15-40μm,25g,CH2Cl2/CH3OH/NH4OH:95/5/0.5)上进行纯化。将纯的级分进行收集并蒸发至干燥,以给出20mg。将该级分用CH3CN吸收,将沉淀物过滤出并在真空下干燥,以提供18mg的最终化合物23(38%产率)。
制备最终产物的总体方案:方法13
中间体O2的合成
使用针对中间体X1(E异构体)所描述的程序来获得中间体O2。
最终化合物21的合成
在室温下,将TiCl3(51.5mL;60.128mmol)逐滴添加到O2(1.3g;3.006mmol)在THF(130mL)中的混合物里。将该混合物在室温下搅拌2h。将该混合物冷却至0℃并且然后用K2CO3粉末进行碱化。将所得泥质混合物通过硅藻土垫过滤,并且将该硅藻土用AcOEt/CH3OH8/2的溶液洗涤。将滤液部分蒸发,以便在过滤白色固体并在真空下在85℃干燥之后给出380mg的最终化合物21(35%产率)。
最终化合物22的合成
将在CH3OH(2mL)+CH3CN(2mL)中的化合物21(118mg,0.331mmol)冷却至10℃。逐滴添加HCl(6M在异丙醇中)(0.16mL,0.993mmol)并且将该混合物在室温下搅拌1h。将该沉淀过滤出,用Et2O和洗涤并且在真空下干燥,以给出109mg的呈HCl盐的最终化合物22(0.76HCl0.81H2O,83%产率)。
制备最终产物的总体方案:方法14
中间体P2的合成
将O2(320mg;0.74mmol)、威尔金森催化剂(137mg;0.148mmol)在THF/CH3OH(50/50)(45mL)中的混合物在10巴压力下在室温下氢化20h。将溶剂在真空下蒸发。将该粗产物通过快速硅胶柱层析(15-40μm;24g)在庚烷/AcOEt80/20中进行纯化,以给出310mg的中间体P2(96%产率)。
最终化合物19的合成
在室温下,将TiCl3(9.5mL;11.049mmol)逐滴添加到P2(0.24g;0.552mmol)在THF(25mL)中的混合物里。将该混合物在室温下搅拌2h。将该混合物冷却至0℃并且然后用K2CO3粉末进行碱化。将所得泥质混合物通过硅藻土垫过滤,并且将该硅藻土用AcOEt/CH3OH8/2的溶液洗涤。将滤液部分蒸发,以便在过滤白色固体并在真空下在85℃干燥之后给出100mg的最终化合物19(50%产率)。
最终化合物20的合成
将在CH3OH(2mL)+CH3CN(4mL)中的化合物19(58mg;0.162mmol)冷却至5℃。逐滴添加HCl(6M在异丙醇中)(81μL;0.486mmol)并且将该混合物在室温下搅拌1h。将该沉淀过滤出,用二异丙醚和洗涤并且在真空下在90℃下干燥,以给出57mg的呈HCl盐的最终化合物20(0.88HCl0.04H2O,89%产率)。
制备最终产物的总体方案:方法15
中间体R2的合成
在0℃下,将偶氮二甲酸二异丙酯(3.8mL;19.107mmol)逐滴添加到A1(4.7g;12.738mmol)、Q2(2.27g;12.738mmol)和PPh3(5g;19.107mmol)在THF(100mL)中的混合物里。将该混合物在室温下搅拌12h。添加EtOAc和水。倾析各层。将有机层用水洗涤,经MgSO4干燥,过滤并蒸发溶剂。将该粗产物通过硅胶柱层析(15-40μm;220g)在庚烷/AcOEt85/15中进行纯化至5.3g的中间体R2(79%产率)。
中间体S2的合成
在室温下,将R2(5.3g;10.015mmol)在THF(80mL)和NH4OH(80mL)中搅拌24h。将该混合物在减压下进行浓缩。将残余物用CH2Cl2吸收,将沉淀(矿物)过滤出并且将滤液在减压下进行浓缩。将该粗产物通过硅胶柱层析(15-40μm;220g)在庚烷/AcOEt85/15中进行纯化,以给出3.65g的中间体S2(78%产率)。
中间体T2的合成
在室温下,将NaH(1.35g;33.88mmol)分部分地添加到烯丙醇(41mL)中。将该混合物在室温下搅拌30min之后,将其在0℃下逐滴添加到S2(4g;8.597mmol)在THF(100mL)中的溶液里。然后将所得混合物在室温下搅拌2h30min并且倾倒入NH4Cl饱和水溶液中。添加EtOAc和NaCl饱和水溶液,将各层进行分离并且将该水层用EtOAc萃取(一次)。合并的有机层经MgSO4干燥,过滤并且将溶剂在减压下去除,以给出一种黄色油。将该粗化合物通过制备型LC(不规则SiOH15-40μm,120g格瑞斯(Grace),液体注射,流动相梯度:庚烷/EtOAc85/15)进行纯化,以给出3.2g的呈黄色油的中间体T2(84%产率)。
中间体U2的合成
将T2(1g;2.26mmol)和氯代二环己基硼烷(1M在己烷中)(904μL;904.013μmol)在无水二氯乙烷(540mL)中的溶液在80℃下和在N2气氛下搅拌1h。将该混合物通过N2鼓泡除气15min,添加格拉布-荷维达催化剂第2代(141.6mg;226μmol),将该混合物再次通过N2鼓泡除气15并且然后在120℃下搅拌16h。再次添加0.25当量的催化剂并且将混合物在120℃下搅拌16h。添加SiliabondDMT(5.9g;3.616mmol)并且将该混合物在室温下搅拌16h。将该混合物通过硅藻土垫过滤并将滤液在真空下进行浓缩,以给出一种黑色油。将该粗化合物通过制备型LC(不规则SiOH15-40μm,80g默克(Merck),流动相:CH2Cl2/AcOEt97/3)进行纯化。将含有产物的级分进行合并,并且将溶剂在真空下去除,以给出335mg的中间体U2(E异构体,36%产率)。
最终化合物25的合成
将铁(0.45g;8.084mmol)添加到U2(0.335g;0.808mmol)在乙酸(24mL)+水(5mL)中的混合物里。将该混合物在50℃下剧烈搅拌5h。
添加CH2Cl2并且将该反应混合物通过硅藻土垫过滤,并且然后用乙酸洗涤。将溶剂在减压下去除。将该粗品通过硅胶柱层析(SiO215-40μm;25g)在CH2Cl2/CH3OH/NH4OH96/4/0.5中进行纯化,以给出154mg的最终化合物25(56%产率)。将该化合物在CH3OH中结晶,过滤并且在真空下在90℃下干燥以给出70mg(25mg产率)。
制备最终产物的总体方案:方法16
中间体V2的合成
使用针对中间体K2所描述的程序用3-丁烯醇作为起始材料来合成中间体V2(3.9g,44%产率)。
中间体W2和X2的合成
将格拉布催化剂第2代(236mg,0.277mmol)添加到V2(1.5g;2.77mmol)在无水CH2Cl2(400mL)中的混合物里。将该混合物在室温下在N2流下搅拌24h。添加SiliabondDMT(3.6g,2.216mmol)并且将该混合物在室温下搅拌12h。将该混合物通过硅藻土过滤,将该硅藻土用CH2Cl2洗涤并蒸发滤液。通过快速层析法在硅胶(15-40μm,80g,CH2Cl2/CH3OH:99.5/0.5)上进行纯化,将纯的级分收集并且蒸发至干燥,以给出0.98g的W2和X2的混合物。将两种异构体通过手性SFC(固定相:CHIRALPAKIC5μm250x20mm,流动相70%CO2、30%CH3OH)进行分离,以给出0.805g的中间体W2(E异构体,57%产率)和0.14g的中间体X2(Z异构体,10%产率)。
最终化合物26的合成
用针对最终化合物18所描述的程序来合成最终化合物26(第一步骤:Y2,0.68g,99%产率;第2步骤:52mg,27%产率)。
最终化合物29的合成
用针对最终化合物18所描述的程序来合成最终化合物29(第一步骤:Z2,0.12g,100%产率;第2步骤:8mg,9%产率)。
制备最终产物的总体方案:方法17
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。







![一种通过不对称[3+3]环化反应合成手性六元碳环嘌呤核苷的方法](https://www.zhichawang.com/images/ui/CN2018109402501/CN2018109402501.jpg)
![一种基于联烯酸酯的[3+2]环加成合成手性五元碳环嘌呤核苷的方法](https://www.zhichawang.com/images/ui/CN2018103279113/CN2018103279113.jpg)




![一种不对称[3+2]环化反应合成手性五元碳环嘌呤核苷的方法](https://www.zhichawang.com/images/ui/CN2017109038084/CN2017109038084.jpg)




动态评分
0.0