

IPC分类号 : C12Q3/00I,C12P19/26I,C12P19/14I,G16C20/10I,G16C20/70I,G01N21/359I,G01N30/02I,G01N21/31I,G01N21/78I







专利摘要
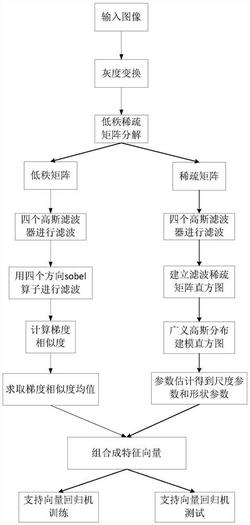
本发明提供一种基于近红外光谱技术的低分子量透明质酸的制备方法,该方法以NIRS技术实现对HA酶解过程的实时监测,通过构建的基于NIRS的LMW‑HAMr预测模型,获取实时的LMW‑HAMr信息,制备得到具有特定分子量的LMW‑HA。本发明的方法能够及时了解酶解进行情况,实现对HAMr的在线实时监测,并能随时根据当前情况作出调整,可以更好的达到生产特定LMW‑HA的目的。
权利要求
1.一种基于近红外光谱技术的低分子量透明质酸的制备方法,其特征在于,所述方法以近红外光谱技术实现对透明质酸酶解过程的实时监测,通过构建基于近红外光谱的低分子量透明质酸相对分子质量预测模型,获取实时的低分子量透明质酸相对分子质量信息,制备得到具有特定分子量的低分子量透明质酸,其中,所述基于近红外光谱的低分子量透明质酸相对分子质量预测模型的构建方法包括:收集低分子量透明质酸样品并测定其相对分子质量;近红外光谱检测,包括采集低分子量透明质酸样品的原始近红外光谱并对采集得到的原始近红外光谱进行预处理;建立近红外光谱与低分子量透明质酸相对分子质量间的关联,建立预测模型;
所述预测模型以近红外全波段10000-4000 cm
所述方法包括以硫酸软骨素酶ABC I酶解透明质酸,在酶解过程中引入近红外光谱技术实时采集和传递近红外光谱信号,并将实时采集到的近红外光谱信号通过基于近红外光谱的低分子量透明质酸相对分子质量预测模型转化为低分子量透明质酸相对分子质量的预测值;
所述预测模型的构建方法中,采用二阶导数25点平滑结合AU处理的方式对样品的近红外光谱进行预处理。
2.根据权利要求1所述的制备方法,其特征在于,所述预测模型的构建方法中,低分子量透明质酸样品的收集包括:
将透明质酸配制成为反应底物溶液,然后向底物溶液中加入硫酸软骨素酶ABC I进行酶解反应,收集反应过程中的透明质酸溶液,保存作为样品待用。
3.根据权利要求1所述的制备方法,其特征在于,所述预测模型的构建方法中,所述低分子量透明质酸样品相对分子质量的测定包括:
选用相对分子质量为参数指标,使用多角度激光散射仪结合凝胶色谱法为测定方法记录收集到的低分子量透明质酸样品的色谱图,计算得到低分子量透明质酸样品的相对分子质量。
4.根据权利要求3所述的制备方法,其特征在于,所述预测模型的构建方法中,色谱条件:Shodex 806M-HQ凝胶柱;0.1 mol/L含0.02%叠氮化钠的氯化钠溶液;流速为0.6 mL/min;检测柱温为35℃;示差折光检测器温度为40℃;进样体积为100 μL;Dn/Dc为0.16。
5.根据权利要求1所述的制备方法,其特征在于,所述预测模型的构建方法中,所述采集低分子量透明质酸样品的原始近红外光谱的步骤包括以AntarisII傅里叶变换近红外光谱仪透射模块采谱,光谱范围为10000-4000 cm
6.根据权利要求1所述的制备方法,其特征在于,所述方法包括:将透明质酸溶解在缓冲体系中,配制反应底物溶液,然后向该溶液中加入硫酸软骨素酶ABC I进行反应,反应过程中实时采集和传递近红外光谱信号,通过基于近红外光谱的低分子量透明质酸相对分子质量预测模型将实时的近红外光谱信号转化为实时的低分子量透明质酸相对分子质量信息,控制反应进程,反应结束后,加热使酶失活,反应液中补加氯化钠,加乙醇沉淀后真空干燥得到特定低分子量透明质酸。
7.根据权利要求6所述的制备方法,其特征在于,所述缓冲体系为Tirs-HCl溶液。
8.根据权利要求7所述的制备方法,其特征在于,所述缓冲体系为50 mol/L Tris-HCl溶液,其制备方法为:称取Tris加入双蒸水500 mL中,用盐酸调节pH为7.0。
9.根据权利要求1所述的制备方法,其特征在于,所述方法包括:将透明质酸溶解在pH值为7.0的Tris-HCl缓冲液中,配制成为反应底物溶液,将此溶液置于25℃温度下,然后向底物溶液中加入硫酸软骨素酶ABC I,通过实时采集和传递近红外光谱信号,通过基于近红外光谱的低分子量透明质酸相对分子质量预测模型将实时的近红外光谱信号转化为实时的低分子量透明质酸相对分子质量信息,控制反应进程;反应结束后,加热使酶失活,通过0.22 μm滤膜过滤除蛋白,向反应液中补加氯化钠使其质量浓度为10g/L,用3-4倍体积乙醇沉淀后真空干燥得到特定低分子量透明质酸。
10.根据权利要求6或9所述的制备方法,其特征在于,所述控制反应进程包括控制硫酸软骨素酶ABC I的加入量、底物溶液浓度、反应时间、pH和温度。
说明书
技术领域
本发明涉及医药领域,具体涉及一种基于近红外光谱技术的低分子量透明质酸的制备方法。
背景技术
公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
透明质酸(HA)又称为玻璃酸,普遍存在于脊椎动物组织间质和一些细菌荚膜中,其相对分子质量(Mr)通常为1×10
HA的Mr检测方法主要有特性黏度法、多角度激光光散射仪结合凝胶色谱法、高效凝胶渗透色谱法等。以上方法均需要先得到纯的透明质酸样品,再将样品溶解于合适的溶液体系中进行检测。且一般利用酶解法制备的透明质酸样品,也需要在样品制备完成之后再利用上述方法对其具体分子量进行检测。检测方法较为繁琐,复杂耗时,在生产过程中也无法实时检测溶液中透明质酸分子量。
目前,制备LMW-HA的方法有很多,主要可分为物理降解法、化学降解法、酶解法、微生物发酵法等。不同的降解方法各有特点,且经不同降解方法得到的LMW-HA在结构和生物活性上也可能存在差异。通过物理因素如加热、微波、超声波、紫外线、机械剪切力等使HA产生降解的方法称为物理降解法,其优点是原理清晰,无需加入其它任何外来物质,方法简便,得到产物热稳定性好。常用的化学降解法包括酸水解法、碱水解法、氧化降解法等。酸水解常使用浓盐酸,碱水解常使用氢氧化钠溶液,氧化降解常用的氧化剂有次氯酸钠、过氧化氢等。化学降解具有成本低、生产规模大的优点,但是缺点也很明显,即降解使用的化学试剂可能会残留,且化学试剂可能会改变HA糖链上的基团或断裂残基的结构,因此影响所制备的LMW-HA的生物活性。酶降解法是制备LMW-HA的首选方法之一,因其反应条件温和副产物少,且具有专一性强、对HA糖链结构不产生破坏等优点。目前降解HA专一性的酶主要有HA酶和硫酸软骨素酶,可通过控制不同的降解条件达到得到不同Mr HA的目的。但由于降解酶的来源比较有限,成本较高,在一定程度上限制了此法的应用。工业上使用工程菌发酵生产HA不仅可以提高产量、降低成本,还可制得具有特定Mr的HA。HA合成酶是HA合成途径中的关键酶,且HA Mr大小可能与HA合成酶的一级结构有关(蒋延超,蒋世云,傅凤鸣,等.透明质酸生物合成途径及基因工程研究进展[J].中国生物工程杂志,2015,35(1):104-110.)。对菌株的HA合成酶的基因进行改造,即可得到能稳定生产LMW-HA的菌株。中国专利CN103993022A中公开了利用同源重组技术将兽疫链球菌中的乳酸脱氢酶(LDH)编码基因进行无缝敲除,获得ldh敲除的重组菌,通过微生物发酵可直接在发酵液中分离获得LMW-HA,成本较低,易于形成规模化工业生产。但是,在发酵过程中底物浓度、发酵温度、搅拌速度、通氧量等因素对所生产的HA的Mr具有影响。
此外,发明人发现酶解法生产LMW-HA时,可能会存在酶制剂产品的功能活性不稳定的问题,导致需要反复调试后续的工艺条件(温度、pH值等)才能达到生产出特定LMW-HA的目的,增加了生产的难度,限制了酶解法在工业化生产上的使用。
近红外光谱(near infrared spectroscopy,NIRS)是在10000-4000cm
此外,发明人发现,虽然NIRS技术可以采集样品的形式为澄清液体、黏稠液体、固体、半固体等,检测时不用改变待测样品的形态,但是其实际应用过程中往往需要克服不同的困难,尤其对于较为复杂的黏稠液体环境,相较于固体形式的单纯检测,需要克服更多更为艰难的问题。比如,将NIRS技术应用于发酵过程的检测时,往往会存在较大的实验误差,造成误差的原因主要在于:发酵液成分复杂,含有大量的菌体、葡萄糖、蛋白质等,复杂的背景信息会对模型产生较大影响;黏稠的发酵液中含有大量的气泡,导致光程发生变化。发酵液处理中醇沉程度不一致导致一级数据不准确,从而引入误差。并且采用NIRS技术建立的模型往往依赖于建立条件的一致性,因而并不具有广泛普适性。
发明内容
如本发明背景技术中所述的,本发明的发明人发现:酶解法生产LMW-HA时可能会存在酶制剂产品的功能活性不稳定的问题,导致需要反复调试后续的工艺条件(温度、pH值等)才能达到生产出特定LMW-HA的目的,增加了生产的难度,限制了酶解法在工业化生产上的使用。
因此,现有的利用酶解法生产特定低Mr的HA的方法只有对酶解的过程进行严格的控制,才能达到理想的生产目的,保证产品的质量和产量。反应罐内酶解进行的情况往往无法直接获取,利用取样分析获取的数据又通常滞后于正在进行的酶解过程,且从取液体酶解反应液到制成可供检测Mr的HA样品的过程往往十分复杂繁琐,这样就增加了生产的难度,也容易降低生产的效率和品质,较为精确的获取具有特定低Mr的HA难以实现。
为了能够及时了解反应罐内酶解进行的情况,并能随时根据反应的实时情况作出调整,就可以更容易地达到生产特定LMW-HA的目的。为此,本发明将NIRS分析技术作为LMW-HA生产过程中的过程分析方法,利用NIR技术可对酶解过程的液体HA Mr进行在线快速测定,省略离线取样制样的繁琐工作,可以达到实时监测反应罐中HA Mr的目的,能大大提高生产的效率和质量,对LMW-HA的生产过程控制和工艺优化具有重要的意义。
具体地,本发明的技术方案如下所述:
本发明提供了一种特定LMW-HA的制备方法,该方法以NIRS技术实现对HA酶解过程的实时监测,通过构建的基于NIRS的LMW-HA Mr预测模型,获取实时的LMW-HA Mr信息,制备得到具有特定Mr的LMW-HA,其中,所述基于NIRS的LMW-HA Mr预测模型的构建方法包括:收集LMW-HA样品并测定其Mr;NIRS检测,包括采集LMW-HA样品的原始NIRS并对采集得到的原始NIRS进行预处理;建立NIRS与LMW-HA Mr间的关联,建立预测模型。
在本发明的实施方式中,所述预测模型以近红外全波段10000-4000cm
在本发明的实施方式中,建模过程中本发明对特征光谱区间进行优化,方法包括选择近红外的全波段10000-4000cm
在本发明的实施方式中,所述特定LMW-HA的制备方法包括以硫酸软骨素酶ABC I酶解HA,在酶解过程中引入NIRS技术实时采集和传递NIRS信号,并将实时采集到的NIRS信号通过基于NIRS的LMW-HA Mr预测模型转化为LMW-HA Mr的预测值。
对NIRS信号的采集和传递方式比如可通过将近红外光纤探头设置在反应体系中,如果应用于生产工艺中,则可将近红外光纤探头设置在反应罐中,对设置方式本领域技术人员可根据实际需要及发酵罐的具体结构进行调整,使实时采集到的NIRS信号能够通过传导光纤及时的传递到控制系统中,所述控制系统也可称为主控制台,比如为计算机。
实时采集及实时传输到控制中心的NIRS信号通过本发明构建的基于NIRS的LMW-HA Mr预测模型(该模型可录入到控制系统中,比如计算机中),转化为实时的LMW-HA Mr信息。同时,根据该实时转化得到的LMW-HA Mr信息反馈到生产进程,通过反馈系统或控制系统,及时控制反应,实现制备特定LMW-HA的目的。
本发明所述的实时转化得到的LMW-HA Mr信息在大规模生产工艺中可与生产过程系统建立实时反馈系统,以实现自动、及时的调整加酶量、底物浓度、反应时间等相关生产条件,最终实现对LMW-HA制备过程的实时最优在线控制,达到大规模生产制备特定LMW-HA的目的。
本发明所述的实时监测系统中至少应含有近红外信号的采集与传递装置,该实时监测系统可以与以下系统或设备中的一种或多种进行交互,比如与控制硫酸软骨素酶ABCI向酶解反应中引入(比如引入到发生酶解应的发酵罐中)的系统或设备,比如与控制HA的加入量和/或控制反应时间以将酶解液中LMW-HA的形成调节到所期望的Mr的设备进行交互。
在本发明的实施方式中,在所述的建模过程中,LMW-HA样品的收集包括:将HA配制成为反应底物溶液,然后向底物溶液中加入硫酸软骨素酶ABC I进行酶解反应,收集反应过程中的HA溶液,保存作为样品待用。
在本发明的实施方式中,所述LMW-HA样品Mr的测定包括:选用Mr为参数指标,使用多角度激光光散射仪结合凝胶色谱法为测定方法记录收集到的LMW-HA样品的色谱图,计算得到LMW-HA样品的Mr;优选地,所述色谱条件:Shodex 806M-HQ凝胶柱;0.1mol/L含0.02%叠氮化钠的氯化钠溶液;流速为0.6mL/min;检测柱温为35℃;示差折光检测器温度为40℃;进样体积为100μL;Dn/Dc为0.16。
在本发明的某些实施方式中,建模过程中,所述LMW-HA样品的收集包括:将HA溶解在pH值为7.0的Tris-HCl缓冲液中,配制成为反应底物溶液,将此溶液置于25℃温度下,然后向底物溶液中加入不同量的硫酸软骨素酶ABC I,控制不同反应时间,使其反应,收集不同加入量以及不同反应时间下的HA溶液,保存在-20℃作为样品待用。
在本发明的某些实施方式中,建模过程中,所述LMW-HA样品Mr的测定包括:
选用Mr为参数指标,使用多角度激光光散射仪结合凝胶色谱法为测定方法;
色谱条件:Shodex 806M-HQ凝胶柱;0.1mol/L含0.02%叠氮化钠的氯化钠溶液;流速为0.6mL/min;检测柱温为35℃;示差折光检测器温度为40℃;进样体积为100μL;Dn/Dc为0.16;
所述测定步骤包括:称取HA样品2.5mg置25mL容量瓶中,加流动相至相近刻度,溶胀过夜,加流动相稀释至刻度,摇匀;用0.22μm微孔滤膜过滤,取续滤液进样测定;记录色谱图,计算得到LMW-HA样品的Mr。
在本发明的实施方式中,建模过程中,所述对LMW-HA样品原始NIRS的采集包括以Antaris II傅里叶变换NIRS仪透射模块采谱,光谱范围为10000-4000cm
在本发明的实施方式中,建模过程中,分别采用AU(标准化)、MC(均值中心化)、MSC(多元散射校正)、SNV(标准归一化)、一阶导数SG平滑和二阶导数SG平滑光谱的预处理方法对原始NIRS进行预处理,采用SVM方法建立Mr模型,以模型评价参数RMSEC、RMSEP、Rc
在本发明的实施方式中,所述的基于NIRS的LMW-HA Mr的预测模型的构建方法包括:从HA酶解过程中获取LMW-HA样品测定其Mr值,并获取LMW-HA样品的原始NIRS,通过RS法(即random sampling法,为随机从样品集中挑选一定的样本作为校正集,剩下的样本作为验证集)划分校正集(Cal)和验证集(Val)样品,对原始NIRS进行二阶导数SG25点平滑结合AU处理,在近红外全波段10000-4000cm
在本发明的实施方式中,所述特定LMW-HA的制备方法包括:将HA溶解在缓冲体系中,配制反应底物溶液,然后向该溶液中加入硫酸软骨素酶ABC I进行反应,反应过程中实时采集和传递的NIRS信号,通过基于NIRS的LMW-HAMr预测模型将实时的NIRS信号转化为实时的LMW-HAMr信息,控制反应进程,反应结束后,加热使酶失活,反应液中补加氯化钠,加乙醇沉淀后真空干燥得到特定LMW-HA。
在本发明的一些实施方式中,所述HA的酶解过程中,所述缓冲体系选自碳酸氢铵溶液、醋酸铵溶液和Tirs-HCl溶液中的一种。优选地,所述缓冲体系为50mol/L Tris-HCl溶液,其制备方法为:称取Tris加入双蒸水500mL中,用盐酸调节pH为7.0。
在本发明的某些实施方式中,所述特定LMW-HA的制备方法包括:将HA溶解在pH值为7.0的Tris-HCl缓冲液中,配制成为反应底物溶液,将此溶液置于25℃温度下,然后向底物溶液中加入硫酸软骨素酶ABC I,通过实时采集和传递的NIRS信号,通过基于NIRS的LMW-HAMr预测模型将实时的NIRS信号转化为实时的LMW-HAMr信息,控制反应进程;反应结束后,加热使酶失活,通过0.22μm滤膜过滤除蛋白,向反应液中补加氯化钠使其质量浓度为10g/L,用3-4倍体积乙醇沉淀后真空干燥得到特定LMW-HA。
在本发明的实施方式中,所述控制反应进程包括控制硫酸软骨素酶ABC I的加入量、底物溶液浓度、反应时间、pH、温度中的一种或多种。
在本发明的实施方式中,本发明所使用的HA的Mr在150万~185万(道尔顿,Da),经酶解后可以得到Mr在1万到180万Da间的HA。为了提高预测结果的准确性,尽可能的避免或减少预测结果与实测结果间的偏差所带来的影响,本发明所述的特定LMW-HA的Mr在6万到178万Da之间,更优选为在10万到178万Da之间,尤其优选在20万到178万Da之间,或者在24万到178万Da之间,或者30万到178万Da之间,或者50万到150万Da之间等。
如无特殊说明,本发明所述两个数值之间的范围包含端值的情况。
本发明将NIRS分析技术作为LMW-HA生产过程中的过程分析方法,利用NIRS技术可对酶解过程的液体HAMr进行在线实时的快速测定,避免了从取液体酶解反应液到制成可供检测Mr的HA样品的复杂繁琐的过程,可以达到实时监测反应罐中HAMr的目的,避免出现反应结束、样品制备完成后再进行检测才发现制备的产品Mr不符合要求的状况。本发明的生产过程中依据建立完善的模型,只需要根据反馈来的NIRS信号在线调整反应条件,就可以达到生产特定Mr透明质酸的要求,而不用再去重新进行实验研究才能更改相应的酶解反应条件。这样也能节约生产成本,大大提高生产的效率和质量,能够较为精确的获取具有特定低Mr的HA。本发明构建的基于NIRS的LMW-HA Mr的预测模型较为准确和可靠,尤其在本发明的制备工艺中具备良好的预测能力。
附图说明
构成本申请的一部分的说明书附图用来提供对本申请的进一步理解,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。以下,结合附图来详细说明本发明的实施方案,其中:
图1为实施例2中的蛋白质含量标准曲线。
图2为实施例2中pH对硫酸软骨素酶ABC I活性的影响曲线。
图3为实施例2中温度对硫酸软骨素酶ABC I活性的影响曲线。
图4为实施例2中硫酸软骨素酶ABC I对不同GAG(糖胺聚糖)的水解作用结果图。
图5为实施例2中金属离子对硫酸软骨素酶ABC I活性的影响结果图。
图6为实施例3中不同缓冲体系对硫酸软骨素酶ABC I活性的影响结果图。
图7为实施例3中氯离子检验结果。
图8为实施例3中HA原料紫外扫描图。
图9为实施例3中LMW-HA样品紫外扫描图。
图10为实施例3中HA原料与LMW-HA样品的紫外扫描对比图。
图11为实施例3中对LMW-HA样品的制备进行单因素考察时,反应时间对HA Mr的影响曲线。
图12为实施例3中对LMW-HA样品的制备进行单因素考察时,酶浓度对HA Mr的影响曲线。
图13为实施例3中对LMW-HA样品的制备进行单因素考察时,底物浓度对HA Mr的影响曲线。
图14为实施例4中用FT-NIR光谱仪采集的55个HA样品的原始NIR光谱图。
图15为实施例4中一阶导数平滑窗口宽度的建模效果图。
图16为实施例4中二阶导数平滑窗口宽度的建模效果图。
图17为实施例4中二阶导数25点平滑结合AU处理后得到的光谱图。
图18为实施例4中HA Mr SVM最佳定量模型图。
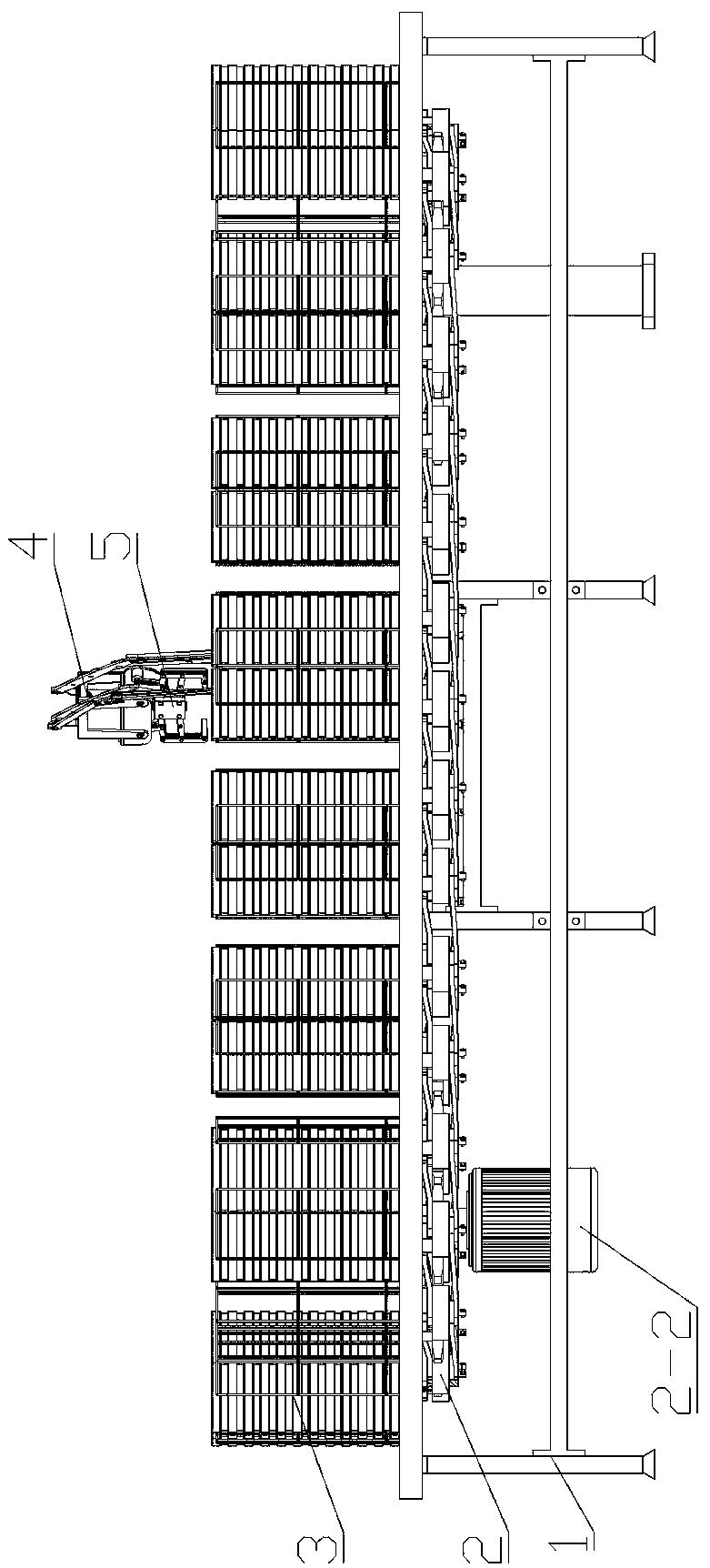
图19为实施例5中LMW-HA制备工艺示意图。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
实施例中使用的仪器及设备:
恒温摇床(ZHWY-100B):海智诚实验设备有限公司;分光光度计(Cary 100):安捷伦公司;真空抽滤装置:天津津腾实验设备有限公司;超净台苏州安泰空气技术有限公司;pH计:梅特勒-托利多仪器有限公司;低温生化培养箱(SHR-1500):上海精宏实验设备有限公司;超声破碎仪:美国SONICS公司;高压灭菌锅(HVE-50):Hirayama公司;分析天平:梅特勒-托利多有限公司;Milli-Q纯水系统:Millipore公司;SHB-III型循环水式多用真空泵:郑州长城科工贸有限公司;台式低温高速离心机(5417R):Eppendorf公司;酶标仪(680):Bio-Rad公司;微量移液器:德国Eppendorf股份公司;AntarisⅡ傅里叶变换近红外光仪:美国Thermo Fisher公司;TW12数显恒温水浴锅:JULABO;Matlab 2015b:Mathworks;RESULTNIRS采集软件:美国Thermo Fisher公司;冷冻干燥机(LYO-0.5):上海东富龙科技股份有限公司;真空干燥箱:德国宾德Binder公司;ASTRA 5.3.4.13软件系统:美国Wyatt公司;多角度激光光散射仪(DAWN EOS):美国Wyatt公司;示差折光检测器:美国Wyatt公司;高效液相色谱仪(600E-996):美国Waters公司;旋转蒸发仪(LABOROTA 4000):德国Heidolph公司。
硫酸软骨素酶ABC I(Chondroitin Sulfate ABC lyase I,ChS ABC lyase I)是在普通变形杆菌(Proteus vulgaris,订购于ATCC)中表达的一种可降解糖胺聚糖的酶,已有研究(J BIOL CHEM,1997,272(14):9123-9130;J.Mol.Biol.(2003)328,623–634)证实该酶为内切酶,可通过β-消除的方式降解硫酸软骨素A-C和HA的1,4-己糖胺键,得到低Mr的硫酸软骨素和HA。目前已有对硫酸软骨素酶ABC I进行重组表达的报道,都是采用大肠杆菌作为载体菌,利用不同的融合载体加入不同的标签以便于提取,经提取后可以达到较高的酶活,证明硫酸软骨素酶ABC I的重组表达是可行的,比如可按照Biochem.J.(2005)386,103–112(Printed in Great Britain);Appl Microbiol Biotechnol(1994)41:39-46;International Journal of Biological Macromolecules 72(2015)6–10中所述重组表达获得硫酸软骨素酶ABC I。本实施例中所述文献将以全文的形式引入本发明。
本实施例采用大肠杆菌(E.coli BL21)作为宿主菌,对构建有His标签的硫酸软骨素酶ABC I进行了重组表达。此菌株表达的硫酸软骨素酶ABC I经提取后对硫酸软骨素和HA均能表现出较高的生物活性。
1.菌株:普通变形杆菌(Proteus vulgaris),大肠杆菌(E.coli BL21)。
2.质粒:pET28a。
3.试剂与材料:LB琼脂培养基,青岛高科园海博生物技术有限公司;酵母提取物、胰蛋白胨,OXOID;氯化钠(分析纯)、三羟甲基氨基甲烷(Tris)、磷酸(分析纯)、盐酸(分析纯),国药集团化学试剂有限公司;咪唑、异丙基硫代半乳糖苷(IPTG)、考马斯亮蓝G-250,北京索莱宝生物有限公司;卡那霉素、镍-琼脂糖凝胶柱美国Sigma公司;乙醇(分析纯),天津市富宇精细化工有限公司;透明质酸(HA),山东福瑞达医药集团有限公司;双蒸水、三蒸水,自制;基因组提取试剂盒,质粒提取试剂盒,限制性内切酶,TaqDNA复制酶。
目的基因的获取:根据Genbank上查到的硫酸软骨素酶ABC I的序列设计引物,PCR扩增获得目的基因,测序;构建工程菌(即可表达硫酸软骨素酶ABC I的E.coli BL21):将目的基因连入质粒,扩增后表达。
4.培养基及常用溶液的配制方法
(1)LB固体培养基:称取LB琼脂培养基1.75g,加入双蒸水50mL,121℃高压蒸汽灭菌20min。
(2)LB液体培养基:称取酵母提取物5g,胰蛋白胨10g,氯化钠10g,加入双蒸水1L,121℃高压蒸汽灭菌20min。
(3)种子培养基:称取酵母提取物0.25g,胰蛋白胨0.5g,氯化钠0.5g,加入双蒸水50mL,121℃高压蒸汽灭菌20min。
(4)100mg/mL卡那霉素溶液:称取卡那霉素1g,加入三蒸水10mL,溶解后于超净台中用0.22μm滤膜过滤,分装于1.5mL Ep管中,置于-20℃保存。
(5)1moL/L IPTG溶液:称取IPTG 0.238g,加入三蒸水1mL,溶解后于超净台中用0.22μm滤膜过滤,分装于1.5mL Ep管中,置于-20℃保存。
(6)平衡缓冲液(5mmol/L咪唑):称取咪唑0.3404g,氯化钠29.22g,Tris 6.057g,加入三蒸水1L,调节pH为7.5,用0.22μm双层滤膜过滤,超声脱气后置于4℃保存。
(7)洗脱缓冲液(80mmol/L咪唑):称取咪唑2.72g,氯化钠14.61g,Tris 3.025g,加入三蒸水500mL,调节pH为7.5,用0.22μm双层滤膜过滤,超声脱气后置于4℃保存。
(8)收集缓冲液(200mmol/L咪唑):称取咪唑6.808g,氯化钠14.61g,Tris 3.025g,加入三蒸水500mL,调节pH为7.5,用0.22μm双层滤膜过滤,超声脱气后置于4℃保存。
(9)考马斯亮蓝检测液:称取考马斯亮蓝G-250 50mg,量取95%乙醇25mL,85%磷酸50mL,加入双蒸水定容至500mL,过滤后置于4℃保存。
硫酸软骨素酶ABC I的制备
1.可表达硫酸软骨素酶ABC I的E.coli BL21菌种的培养
(1)菌种纯化:将冻存于-80℃的可表达硫酸软骨素酶ABC I的E.coli BL21菌种置于冰上使其缓慢冻融。在50mL固体培养基中加入100mg/mL的卡那霉素溶液50μL。铺两个平板,待培养基凝固后,用接种环沾取菌种,在固体培养基上用三线法划线。划好线后的培养皿用封口膜封口,低温生化培养箱中37℃倒置培养过夜。
(2)菌种活化:在50mL种子培养基中加入100mg/mL的卡那霉素溶液50μL,在平板中挑一个单菌落至种子培养基中,恒温摇床中37℃、225r/min培养过夜。
(3)扩大培养:在1L培养基中加入100mg/mL的卡那霉素溶液1000μL,加入活化后的菌液约10mL,恒温摇床中37℃、225r/min培养至其OD值为0.6~0.8,大约3h。
(4)诱导培养:在1L培养基中加入1moL/L的IPTG溶液200μL,恒温摇床中20℃、225r/min培养20h。
2.菌体的处理
(1)诱导培养结束后,用台式低温高速离心机将所得菌液在8000r/min条件下离心20min。弃去上清液,用少量平衡缓冲液重悬沉淀。
(2)用超声破碎仪破碎重悬菌液,参数设置为作3s,间歇5s,工作时间为40min,工作功率150兆,破碎至菌液澄清透明。
(3)用台式低温高速离心机将破碎后的菌液在4℃、8000r/min条件下离心40min。弃去沉淀,保留上清液,并用0.22μm滤膜上清液。
3.硫酸软骨素酶ABC I的纯化和保存
硫酸软骨素酶ABC I的纯化使用镍-琼脂糖凝胶柱进行,步骤如下:
(1)平衡:4℃下先使用三蒸水冲洗柱子至柱子中无乙醇,约10个柱体积,流速为0.5mL/min。再用平衡缓冲液(5mmol/L咪唑)平衡柱子,流速为0.5mL/min。
(2)上样:过滤后的上清液,4℃下上样到镍-琼脂糖凝胶柱上,流速为0.5mL/min。
(3)再平衡:4℃下使用流速为0.5mL/min的平衡缓冲液(5mmol/L咪唑)1-2个柱体积来平衡柱子。
(4)洗脱:4℃下使用洗脱缓冲液(80mmol/L咪唑)冲洗柱子,流速为0.5mL/min,用考马斯亮蓝检测洗脱液,观察颜色由亮蓝色至浅蓝色,表示基本再无蛋白被洗脱下来。
(5)收集:4℃下使用收集缓冲液(200mmol/L咪唑)洗脱目的蛋白,流速0.5mL/min,马斯亮蓝检测蓝色出现时接样品,直至蓝色变浅,停止收样。继续用收集缓冲液冲洗柱子约30min。换用三蒸水冲洗柱子30min后,再用20%乙醇将柱子保存在4℃。
(6)酶的保存:将收集到的考马斯亮蓝检测蓝色较深的几组样品混合,得到纯酶液。取收集缓冲液和甘油按1:1的比例配置50%甘油溶液。在2mL Ep管中加入50%甘油溶液200μL,纯酶液800μL,充分混合均匀后置于保存在-80℃。
1.硫酸软骨素酶ABC I的酶活力测定方法
酶活力单位定义:在本实施例中,酶的活力单位U定义为:在25℃、pH7.0的条件下,催化0.2%HA溶液反应,每1min在波长232nm处引起吸光度升高0.001的酶量。
酶活力的测定:取一定量的0.2%HA溶液,置于2mL Ep管中,加入一定体积的硫酸软骨素ABC I酶液,在25℃条件下进行反应后,立即加热煮沸5min终止反应。用紫外分光光度计在232nm处检测光吸收值(A232)。
酶活力的计算公式,如下:
式中:ΔA232指反应前后吸光度的变化量;t指反应的时间(min);Vt指反应的总体积(mL);Vs指反应中加入酶液的体积(μL)。
在研究酶学性质时,用相对酶活(%)来表示各因素对硫酸软骨素酶ABC I活力的影响。其中相对酶活指在单因素酶学性质实验中,本实施例任意指定其中一水平的酶活力为100%时余下各水平酶活力与其的比值。使用相对酶活可以使实验结果的表达更直观清晰。
2.蛋白质含量的测定
采用BCA试剂盒法测定蛋白质含量。具体操作步骤为:
(1)配制工作液:根据标准品和样品数量,按BCA试剂A:BCA试剂B体积为5:1配制适量BCA工作液,充分混匀,现配现用。
(2)稀释标准品:现有标准品浓度为5mg/mL,稀释使其终浓度为0.5mg/mL。
(3)绘制标准曲线:将稀释后的标准品按0、1、2、4、8、12、16、20μL加到96孔板的样品孔中,用PBS溶液将每孔补足至20μL。向各孔加入BCA工作液200μL,37℃放置30-60min。冷却至室温后在酶标仪上562nm处进行比色,以空白单位作为对照组,标准品蛋白含量作为横坐标,吸光值作为纵坐标,绘制出蛋白质含量的标准曲线,如图1。
(4)样品蛋白质含量的测定:加适量体积待测样品溶液到96孔板的样品孔中,用PBS溶液将样品孔补足至20μL。向其中加入BCA工作液200μL,37℃放置30-60min。冷却至室温后,以空白为对照,在酶标仪上562nm处比色,根据样品的吸光值从标准曲线上查出样品的蛋白质含量。
3.硫酸软骨素酶ABC I部分酶学性质的研究
最适反应pH的测定:用缓冲液配制不同pH值的HA底物溶液,使反应缓冲体系的pH分别为4.0、5.0、6.0、7.0和8.0,分别加入等体积的硫酸软骨素ABC I,其余反应条件均相同,测定其酶活力,考察不同pH值对酶反应活力的影响。
最适反应温度的测定:
分别在20、25、30、40、50℃条件下进行酶反应,加入等体积的硫酸软骨素ABC I,其余反应条件均相同,测定其酶活力,考察不同温度对酶反应活力的影响。
酶的底物专一性研究:分别配置浓度为0.2%的硫酸软骨素、HA、肝素、硫酸皮肤素、壳聚糖溶液,加入等体积的硫酸软骨素ABC I,其余反应条件均相同,测定其酶活力,考察硫酸软骨素酶ABC I的底物专一性。
金属离子对酶反应的影响:
用缓冲液配制相同浓度的K
4.实验结果
(1)硫酸软骨素酶ABC I的获取及酶活力的测定:每次实验时配置培养基2L,经过镍-琼脂糖凝胶柱初步纯化后可获得具有酶活力的硫酸软骨素酶ABC I酶液约10mL左右,加入10%的甘油混合均匀后保存在-80℃,取用时放置在冰盒中待其融化后使用。
本实施例中所添加的硫酸软骨素酶ABC I其总酶活为1.38×10
(2)硫酸软骨素酶ABC I部分酶学性质的研究
最适反应pH的测定:
pH对酶活力的影响如图2所示。由实验结果可知,当pH的范围在4.0~7.0时,随着pH的升高,硫酸软骨素酶ABC I的活性也随之升高;当pH值为7.0时,硫酸软骨素酶ABC I的活性最好;当pH大于7.0时,对硫酸软骨素酶ABC I活性影响很大,pH为8时酶活性下降十分迅速。因此本实施例选择硫酸软骨素酶ABC I的最适反应pH为7.0。
最适反应温度的测定:
温度对酶活力的影响如图3所示。由实验结果可知,当温度的范围在20~30℃时对硫酸软骨素酶ABC I的活性影响不大,酶活可保持在较高水平范围内;当温度大于30℃后,硫酸软骨素酶ABC I的活性明显下降。说明温度对硫酸软骨素酶ABC I的影响十分明显,硫酸软骨素酶ABC I可以在一定温度范围内保持较高的酶活力,一旦超出最适的温度范围,酶活力会急剧下降。通过实验选择硫酸软骨素酶ABC I最适的反应温度为20~30℃。
酶的底物专一性研究:
酶的底物专一性研究如图4所示。由实验结果可知,硫酸软骨素酶ABC I对硫酸皮肤素、硫酸软骨素、HA均有较好的降解作用,可测得较高的酶活力;而对肝素和壳聚糖的降解作用不明显,酶活性较低,几乎没有反应。
金属离子对酶反应的影响:
不同金属离子对酶活力的影响如图5所示。由实验结果可知,除Ba
本发明实施例1和实施例2制备了硫酸软骨素酶ABC I并测定了所制得的硫酸软骨素酶ABC I的酶活力,并对其部分酶学性质进行了研究。
实验对IPTG诱导培养的菌体破碎后离心,取其上清液过镍-琼脂糖凝胶柱进行初步纯化,获得具有酶活力的硫酸软骨素酶ABC I。得到的酶以液体的形式加入10%甘油保存在-80℃。经过检测所制的硫酸软骨素酶ABC I具有较高的酶活,其比活力为5.4×10
通过实验检测了硫酸软骨素酶ABC I最适pH和最适温度。由实验结果可知,实施例1制备的硫酸软骨素酶ABC I反应的最适pH为7.0,反应的最适温度范围为20-30℃,在后续实验中我们选取了25℃作为最适反应温度。
对硫酸软骨素酶ABC I的底物专一性研究显示,该酶对一部分糖胺聚糖有十分良好的生物活性如硫酸软骨素、硫酸皮肤素、HA等,但对肝素、壳聚糖则几乎没有降解作用。通过实验还发现除了Ba
1.实施例用品
试剂与材料
透明质酸(HA),山东福瑞达医药集团有限公司;乙醇(分析纯),天津市富宇精细化工有限公司;碳酸氢铵(分析纯)、乙酸铵(分析纯)、三羟甲基氨基甲烷(Tris)、氯化钠(分析纯)、硝酸银(化学纯)、冰醋酸(分析纯)、硝酸(分析纯)、盐酸(分析纯),国药集团化学试剂有限公司;硫酸软骨素酶ABC I(实施例1制备)、双蒸水、三蒸水,自制。
常用溶液的配制方法
(1)50mol/L Tris-HCl溶液:称取Tris 3.028g,加入双蒸水500mL,用盐酸调节pH为7.0。
(2)5mol/L醋酸铵溶液:称取醋酸铵0.19g,加入双蒸水500mL,用冰醋酸调节pH为7.0。
(3)50mol/L碳酸氢铵溶液:称取碳酸氢铵1.976g,加入双蒸水500mL。
(4)2mg/mL HA溶液:称取HA 1g,加入Tris-HCl缓冲溶液500mL,搅拌溶解后置于4℃保存。
(5)0.1mol/L硝酸银溶液:称取硝酸银0.17g,先溶解于适量双蒸水中,再定容至10mL的棕色容量瓶中,摇匀。置于暗处保存。
(6)0.1mol/L氯化钠溶液(含0.02%叠氮化钠):称取氯化钠5.844g,叠氮化钠0.8g,加入三蒸水4L,搅拌溶解,用0.22μm双层滤膜过滤。
2.LMW-HA样品制备方法的研究
最适反应缓冲体系的确定:分别用水、碳酸氢铵、醋酸铵、Tirs-HCl等不同缓冲体系配置浓度相同的HA底物溶液,加入同等体积的酶液,相同条件下进行反应,反应结束后测定酶活力,考察确定最适缓冲体系。
氯离子检验:根据已经确定的最适反应缓冲体系,想要制得符合预期的可供检测Mr的LMW-HA样品,则需要除去样品制备过程中产生的氯化钠。本实施例采用乙醇沉淀的方法制备样品,采用硝酸银溶液检验样品中的杂质氯化钠是否已去除干净。
具体操作步骤为:分别取少量HA原料和HA样品溶液3mL于试管中,各滴入稀硝酸溶液5滴后,将少量硝酸银溶液滴入到试管中,轻轻震荡,观察试管中是否有白色沉淀生产。
紫外光谱扫描:分别量取反应前的HA原料溶液和反应后的HA样品溶液各1mL,其浓度均为2mg/mL。在190~300nm波长范围内进行紫外全波长扫描。
LMW-HA样品分子量的检测:
选用Mr(重均分子量Mw)为本实施例的参数指标,使用多角度激光光散射仪结合凝胶色谱法为测定方法。
色谱条件:Shodex 806M-HQ凝胶柱;0.1mol/L氯化钠溶液(含0.02%叠氮化钠);流速为0.6mL/min;检测柱温为35℃;示差折光检测器温度为40℃;进样体积为100μL;Dn/Dc为0.16。
操作步骤:称取HA样品2.5mg置25mL容量瓶中,加流动相至相近刻度,溶胀过夜,加流动相稀释至刻度,摇匀。用0.22μm微孔滤膜过滤,取续滤液进样测定。记录色谱图,采用专用软件处理数据并计算Mr。
3.LMW-HA样品制备的单因素实验
将一定量的HA溶解在pH值为7.0的Tris-HCl缓冲液中,配制成为反应底物溶液,将此溶液置于25℃温度下,然后向底物溶液中加入不同量的硫酸软骨素酶ABC I,控制不同反应时间,使其反应。反应结束后,加热使酶失活,通过0.22μm滤膜过滤除蛋白,取少量反应液以供近红外建模。剩余反应液补加至1%氯化钠(即向反应液中补加氯化钠使其质量浓度为10g/L),用3~4倍体积乙醇沉淀后真空干燥得到产品。使用多角度激光光散射仪结合凝胶色谱法测定制备的LMW-HA样品的Mr,考察反应时间、加酶量和底物浓度等各因素对HA样品Mr的影响。
反应时间对HA Mr的影响:取pH值为7.0,浓度为2mg/mL HA溶液25mL,将溶液在25℃水浴锅中预热25min,加入硫酸软骨素酶ABC I使酶浓度为5×10
酶浓度对HA Mr的影响:取pH值为7.0,浓度为2mg/mL HA溶液25mL,将溶液在25℃水浴锅中预热25min,加入硫酸软骨素酶ABC I使酶浓度分别为2×10
底物浓度对HA Mr的影响:取pH值为7.0,底物浓度分别为1、1.5、2、2.5、5mg/mL HA溶液各25mL,将溶液在25℃水浴锅中预热25min,加入硫酸软骨素酶ABC I使酶浓度为5×10
LMW-HA样品制备的多因素正交实验:在硫酸软骨素酶ABC I降解HA的反应中,本实施例发现影响降解的主要因素有反应时间(因素A)、加酶量(因素B)和底物浓度(因素C),于是每个因素选择3个水平(表1)进行正交实验设计。
表1正交实验设计
4.实验结果
(1)LMW-HA样品制备方法的建立
最适反应缓冲体系的确定:不同缓冲体系对硫酸软骨素酶ABC I的影响如图6所示。由实验结果可知,硫酸软骨素酶ABC I在Tris-HCl缓冲体系和醋酸铵缓冲体系中酶活较高,但由于考虑到后续样品的制备,选择Tris-HCl缓冲体系作为最适缓冲体系。
(2)氯离子检验
氯离子检验结果如图7所示。由实验结果可知,HA原料与制得的LMW-HA样品的检验结果相同,在加入硝酸银溶液后均没有白色沉淀生成,因此可以认为通过一系列实验操作后,所制得的LMW-HA样品中不含有氯化钠,符合制备样品的预期目的。
(3)紫外扫描光谱
紫外扫描光谱结果如图8、图9所示,由HA原料和LMW-HA两者对比图如图10所示,可知所制得LMW-HA样品光谱除在220-240nm范围内与原料光谱有较明显不同外,其余部分均与原料光谱大致相同,尤其在260、280nm附近曲线平坦,说明样品中几乎不含蛋白质和核酸,可以认为酶解过程中添加的酶蛋白经过一系列实验操作后已被除去,符合制备样品的预期目的。
(4)LMW-HA样品制备方法的确定
通过实验确定LMW-HA样品的制备方法为:将一定量的HA溶解在pH值为7.0的Tris-HCl缓冲液中,配制成为反应底物溶液,将此溶液置于25℃温度下,然后向底物溶液中加入不同量的硫酸软骨素酶ABC I,控制不同反应时间,使其反应。反应结束后,加热使酶失活,通过0.22μm滤膜过滤,取少量反应液以供近红外建模。剩余反应液补加氯化钠至含量为1%(即向反应液中补加氯化钠使其质量浓度为10g/L),用3~4倍体积乙醇沉淀后真空干燥得到产品。
5.LMW-HA样品制备的单因素实验的结果
反应时间对HA Mr的影响实验:
其结果如图11所示。在酶解反应开始时,HA的Mr降低十分迅速;随着酶解时间不断的延长,HA的降解速度开始减缓。由实验结果还可以看出,控制反应时间可制得不同Mr的HA。
酶浓度对HA Mr的影响实验:
结果如图12所示。由实验结果可知,随着酶浓度的不断增加,在相同的反应时间下,HA的最终Mr不断减小。且当加入的酶浓度小于6×10
底物浓度对HA Mr的影响:
结果如图13所示。由实验结果可知,在其余反应条件一致的情况下,随着HA底物浓度不断升高,HAMr不断增大。推测是由于底物浓度增加时,HA溶液的黏度会因此增大而不利于反应的进行。因此综合考虑,底物浓度可以控制在2mg/mL左右。
6.LMW-HA样品制备的多因素正交实验结果
在LMW-HA样品制备的单因素实验结果的基础上,以加入酶的浓度、反应时间和底物浓度为正交因素进行实验设计,正交实验方案与结果见表2。根据R的大小分析,以上各因素对硫酸软骨素酶ABC I酶解HA反应的影响大小依次为:底物浓度、酶浓度、时间。根据各因素各水平所对应指标结果的平均值k1、k2、k3的大小可以确定各因素较优水平组合为A3B3C1,即反应条件为酶浓度为6×10
表2正交实验设计方案与结果
Mr的测量使用多角度激光光散射仪结合凝胶色谱法为测定方法。为了考察不同酶解条件与制得HA Mr之间的关系,本实施例设计了单因素实验和多因素正交实验。由于实施例2中考察了酶的最适pH和温度,为了保证足够的酶活性,在设计单因素实验时保证其在最适pH和温度条件下,主要考察了酶浓度、反应时间、底物浓度3个因素对酶解反应的影响。然后又设计了正交实验,根据R的大小分析,以上3个因素对硫酸软骨素酶ABC I酶解HA反应的影响大小依次为:底物浓度、酶浓度、反应时间。说明在利用硫酸软骨素酶ABC I降解HA时,底物浓度对反应的影响最大。单因素实验中还发现随着底物浓度的增加,制备的LMW-HA的Mr也会不断增大。认为可能是由于底物的浓度增加时,溶液的黏度也在增加从而导致酶活性降低。另外,根据各因素各水平所对应指标结果的平均值k1、k2、k3的大小可以确定各因素较优水平组合为A3B3C1,即反应条件为酶浓度为6×10
HA的Mr检测方法主要有特性黏度法、多角度激光光散射仪结合凝胶色谱法、高效凝胶渗透色谱法等。这些方法的操作过程较为繁琐,无法达到快速准确检测HAMr的目的。为了在LMW-HA制备过程中应用NIRS法实现LMW-HAMr的在线监测与基于此的在线控制,本发明研究建立了NIRS与LMW-HA Mr关系的模型。本发明首先检测了HA样品的Mr作为一级数据,采集了样品对应的反应过程中的反应液的NIRS,通过研究选择光谱最佳的预处理方法、优化光谱特征区间,建立了基于NIRS的LMW-HA Mr模型,并对模型的预测能力进行了分析检验,为实现在反应过程中能实时的检测HA的Mr奠定了基础。
1.试剂与材料
LMW-HA样品(实施例3中制备);双蒸水,自制。
2.实验方法
(1)HA样品的收集和Mr的测定
实施例3中部分反应后的HA溶液,保存在-20℃待用,并测定了该溶液对应制得的LMW-HA样品的Mr。Mr测定方法同实施例3中“LMW-HA样品分子量的检测”中所述,共得到了55份样品。
(2)HA样品NIRS的采集
用AntarisⅡ傅里叶变换NIRS仪透射模块采谱,光谱范围为10000-4000cm-1,扫描次数:32次;分辨率:8cm-1;以空气为背景参比,为了降低信噪比,每1h扣除一次背景。随机将样品注入一个光程为1mm的石英比色皿中,利用控温附件将采谱温度控制在25℃(在测样之前将样品置于25℃水浴锅中预热30min),每个样品采谱3次,取其平均得到样品的原始NIRS图。
(3)校正集、验证集样品的划分
样品集根据random sampling(RS)法进行划分。随机从样品集中挑选一定的样本作为校正集,剩下的样本作为验证集。
(4)预处理方法的选择
为了提高光谱的质量,去除不相关的信息,本实施例中分别采用了AU、MC、MSC、SNV、一阶导数SG平滑和二阶导数SG平滑光谱预处理方法对原始光谱进行预处理,采用支持向量机(Support Vector Machine,SVM)方法建立了Mr模型,以模型评价参数RMSEC、RMSEP、Rc
式中:yi,reference指参考方法测定的样品真值;
yi,predicted指NIRS模型计算得到的样品预值;
指参考方法测定的样品真值的平均值;
n指集合中的样品数。
(5)特征光谱区间的优化
筛选了预处理方法之后,本发明在最佳预处理方法的基础上选择不同特征光谱区间考察其对HA Mr模型建立的影响,对建立的模型进行进一步优化。
由于本实施例中用来采集NIRS图的样品是以溶液的形式存在的,因此考虑利用水光谱组学来考察特征光谱区间对HA Mr模型的影响。水光谱组学是指以生命体系中的水作为研究对象,利用光谱技术监测在不同环境下水分子结构的变化,从分子水平上反映体系中水分子与其他分子之间的相互作用或者反映水在生命体系中的功能。水作为一种天然的生物基质,具有很强的氢键作用,结合系统与环境中的物理或化学变化则会改变其吸光度模式,因此可以利用光谱变化反应系统中其他分子和含量和结构变化。于是本实施例选取了水的一级倍频7700-6250cm
此外,本实施例中还选取了HA的特征光谱区间6887-5411、5123-4348、9091-7331cm
分别在以上两个特征区间建立模型并与全波段下建立的模型做比较,考察光谱区间对HA Mr模型建立的影响,选取最佳建模光谱区间。
(6)模型的建立与分析
因为模型的预测能力受建模方法的影响较大,所以在建立模型的过程中,只有选择了恰当的建模方法才能达到预期的建模效果。本实施例采用SVM建立模型。在通过光谱预处理最佳方法的选择、特征光谱区间的优化建立了最佳模型之后,通常采用代入验证集样品的方法对建模的效果进行检验,利用配对t检验的方法考察模型的预测值与一级分析方法之间是否存在显著性差异。
3实验结果
(1)HA Mr的测量结果
使用多角度激光光散射仪结合凝胶色谱法测定收集的对应LMW-HA样品的Mr,测定结果如表3所示。
表3 HA样品Mr测定结果
由表3可知,得到的HA Mr具有一定的范围和梯度,可以用来建立近红外模型。
(2)HA样品的NIRS图
图14为用FT-NIR光谱仪采集的55个HA样品的原始NIRS图。
从图14可以观察到,HA样品原始NIRS图趋势很相似,不同样品光谱差异不明显,光谱间相似性很高,所以可能需要采取一些化学计量学方法等光谱分析手段来增强光谱的分辨率,减少或除去一些其他不必要的因素对光谱质量的影响。
(3)光谱预处理方法的选择
NIRS图中通常会含有一些如光的散射、杂散光、与及仪器响应等与待测样品性质无关的干扰因素。为提高光谱的质量,消除或减弱因物理因素、化学因素或仪器噪声等对光谱质量的影响,往往会采取特定的化学计量学方法预处理得到的光谱。
本实施例中共对比了6种预处理方法:AU、MC、SNV、MSC、一阶导数、二阶导数。其中由于导数会使光谱的信噪比降低,因此常和SG平滑结合保证消除基线漂移、强化谱带特征的同时提高信噪比。平滑处理结合导数的过程中,平滑窗口的宽度对后续建立的模型质量有非常大的影响。如果选择窗口的宽度太小,就没有办法滤除无用信息,窗口的宽度选择太大,则容易使一些有效信息被过滤掉。所以在平滑处理结合一阶导数和二阶导数的过程中要先优化选择窗口宽度,优化结果如图15和图16。从图中可以看出一阶导数结合SG13点平滑时建模效果最好,为最佳平滑窗口宽度。二阶导数最佳平滑窗口宽度为25点平滑,此时得到的模型的RMSEC和RMSEP值最低,模型结果最好。
表4不同预处理方法的建模效果
选择不同预处理方法对光谱进行处理后得到的建模结果如表4所示,对于Mr模型,光谱经过二阶导数25点平滑结合AU处理后得到的模型的结果最好,得到最佳预处理光谱图如图17所示。
(4)特征光谱区间优化的选择结果
特征光谱区间优化的选择结果如表5所示。通过数据分析,本发明发现在全波段下建立的模型要远远优于所选取的其它波段下建立的模型。因此依旧选取全波段光谱区间作为最佳的建模光谱区间。
表5特征光谱区间优化的选择结果
(5)模型建立
经过预处理光谱、优化选择了光谱的特征区间之后,本发明建立了最佳的NIRS用于HA Mr测定的数学分析模型,如图18所示。通过RS法划分样品集,对光谱进行二阶导数SG25点平滑处理,在近红外全波段建模,得到的模型的RMSEC、RMSEP、Rc
(6)模型的评价
为了验证HA Mr SVM模型的预测能力,本实施例将由参考方法多角度激光光散射仪测得的数据与建立的NIR定量模型预测得到的Mr进行配对t检验。表6、7分别为参考方法测得的数据与NIRS法预测得到的数据及其配对结果。通过配对t检验显示两组数据的平均值和SD值比较接近,且P值为
一种基于近红外光谱技术的低分子量透明质酸的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




















动态评分
0.0