

IPC分类号 : C07J17/00,C07J21/00,A61K31/7048,A61K31/58,A61K31/585,A61K31/7004,A61K31/7028,A61P35/00







专利摘要
本发明实施例公开了一种抗肿瘤化合物、其提取方法及其应用,该化合物具有通式(Ⅰ)或通式(Ⅱ)及其光学异构体的结构;本发明化合物及其组合物能有效地抑制激素非依赖性前列腺癌细胞、不同种类肾癌细胞以及人恶性黑色素瘤细胞的生长,提示本发明化合物及其组合物可作为治疗肿瘤的药物,具有良好的研究开发前景。
权利要求
1.如下式所示的抗肿瘤化合物或其光学异构体:
上述各化合物从毛酸浆的干燥茎叶和/或果实中分离而得。
2.一种如权利要求1所述的化合物的提取方法,其特征在于,包括以下步骤:
采用毛酸浆的干燥茎叶和/或果实为原料,用体积分数为1-99%的乙醇或甲醇的水溶液回流提取1-3次,每次提取1-48小时,合并得到提取液,将提取液去除溶剂得到总浸膏;
将总浸膏用有机溶剂进行萃取,得到有机萃取液,将有机萃取液浓缩后得到有机溶剂层浸膏;所述有机溶剂为甲醇、乙醇、丙酮、乙酸乙酯、氯仿、石油醚及正丁醇中的一种或几种;
将有机溶剂层浸膏通过色谱分离法进行分离,得到如权利要求1所述的化合物。
3.如权利要求1所述的化合物在制备抗肿瘤药物中的应用。
4.如权利要求3所述的应用,其特征在于,所述肿瘤包括:肺癌、支气管癌、肝癌、卵巢癌、子宫颈癌、膀胱癌、睾丸癌、肾细胞癌、胰腺癌、胃癌、骨癌、食道癌、结肠癌、胆管癌、前列腺癌、绒毛膜癌、黑色素瘤、胶质细胞瘤、神经纤维瘤、纤维肉瘤或淋巴管瘤。
5.一种包含如权利要求1所述化合物的植物毛酸浆提取物。
6.如权利要求5所述的植物毛酸浆提取物在制备抗肿瘤药物中的应用,其特征在于,所述肿瘤包括:肺癌、支气管癌、肝癌、卵巢癌、子宫颈癌、膀胱癌、睾丸癌、肾细胞癌、胰腺癌、胃癌、骨癌、食道癌、结肠癌、胆管癌、前列腺癌、绒毛膜癌、黑色素瘤、胶质细胞瘤、神经纤维瘤、纤维肉瘤或淋巴管瘤。
7.一种药物组合物,其特征在于,包括权利要求1所述化合物中的一种或几种。
8.一种药物组合物,其特征在于,包含权利要求1所述的化合物或其药学上可接受的盐,和药学上可接受的载体和/或赋形剂。
9.如权利要求7或8所述的组合物在制备抗肿瘤药物中的应用,其特征在于,所述肿瘤包括:肺癌、支气管癌、肝癌、卵巢癌、子宫颈癌、膀胱癌、睾丸癌、肾细胞癌、胰腺癌、胃癌、骨癌、食道癌、结肠癌、胆管癌、前列腺癌、绒毛膜癌、黑色素瘤、胶质细胞瘤、神经纤维瘤、纤维肉瘤或淋巴管瘤。
说明书
技术领域
本发明涉及一种化合物,特别地涉及从毛酸浆中提取的天然化合物、该化合物的提取方法、包含该化合物的药物组合物以及该化合物和该药物组合物在制备抗肿瘤药物中的用途。
背景技术
毛酸浆Physalis pubescens L.为茄科酸浆属一年生草本植物,在徐国钧主编的《中国药材学》中记载了毛酸浆的带宿萼果实作为灯笼草药用。毛酸浆性味酸平,入肺经,具有清热解毒、清咽利喉、利尿止血之功效。民间用于治疗急性咽炎效果显著。(徐国均.中国药材学.下册[M].北京:中国医药科技出版社。1996:1161-1162.)其传统功效与中医治疗前列腺癌所采用的“行瘀散结,通利水道”的方法恰好一致。
对酸浆属植物的化学研究结果表明,其中主要含有甾体、生物碱、黄酮、萜类等成分(邱莉.酸浆化学成分及其生物活性研究.沈阳药科大学博士学位论文.2007年.导师:邱峰)。酸浆属植物中含有的甾体类成分以withanolide类型为主,是一种含有28个碳原子的麦角甾内酯类化合物,具有抗肿瘤、杀寄生虫、抗菌、抗炎、免疫调节等多方面的药理活性。有研究报道,withanolide类化合物具有抗多种肿瘤细胞的作用:Physalins A和PhysalinsB可通过下调雄激素受体的表达和激活细胞凋亡从而有效抑制雄激素非依赖性前列腺癌细胞的生长(Han H.Y.;Qiu L.;Wang X.H.;et al.Biol.Pharm.Bull.2011,34(10)1584—1588);withanolide类化合物可以选择性抑制肾上腺皮质癌细胞的生长(Subramanian,C.;Zhang,H.P.;Gallagher,R.;et al.World J.Surg.2014,38,1343–1352.);Physalin A可以诱导纤维素肉瘤细胞以及人恶性黑色素瘤细胞的凋亡和自噬(He H.;Zang L.H.;FengY.S.;et al.Physalin A Induces Apoptotic Cell Death and Protective AutophagyinHT1080Human Fibrosarcoma Cells.Journal of Natural Products,2013,76,880–888;HeH.;Feng Y.S.;Zang L.H.;et al.Nitric oxide induces apoptosis and autophagy;autophagy down-regulates NO synthesis in physalin A-treated A375-S2humanmelanoma cells.Food and Chemical Toxicology,2014,71,128–135)等。
但迄今为止,国内外对毛酸浆的活性成分报道甚少,为探索这一具有药用价值的植物(即毛酸浆)是否具有抗肿瘤活性成分,发明人对毛酸浆中具有抗肿瘤的活性成分进行了研究。
发明内容
本发明首要目的在于提供毛酸浆植物中具有抗肿瘤的化合物,其提取方法及其在制备抗肿瘤药物中的应用。
本发明的又一目的在于提供包括上述化合物的组合物及其在制备抗肿瘤药物中的应用。
本发明的目的通过下述技术方案实现:
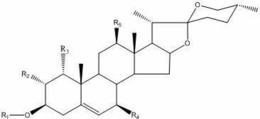
本发明第一方面提供了一种抗肿瘤化合物,具有通式(Ⅰ)、通式(Ⅱ)或两者光学异构体的结构:
其中,通式(Ⅰ)中,2-3位、5-6位为单键或双键;R1为OH或羰基;R2为H、OH、或基团D、E;R3为H或OH;R4为OH或R4与R5结合为基团B;R5为H,Cl或R5与R4结合为基团B;R6为H或基团C;R7为H或基团C;R8为OH、基团A、或R8与R9结合为基团B;R9为OH或R9与R8结合为基团B;R10为OH,羰基或基团A;
所述基团A、基团B、基团C、基团D、及基团E分别为:
在本发明第一方面的一些优选实施方式中,所述的化合物包括:
Physapubside A(1)、Physapubside B(2)、Physapubside C(3)、Physapubside D(4)、Physapubescin E(5)、Physapubescin F(6)、Physapubescin G(7)、Physapubescin H(8)、Physapubescin I(9)、Physapubescin J(10)、Physapubescin K(11)、PhysapubescinL(12)、Physapubescin M(13)、Physapubescin N(14)、Physapubescin O(15)、Physapubescin P(16)、Physapubescin Q(17)、Physapubescin R(18)中的任一种,自化合物1-18的结构式分别为:
上述化合物的制备方法包括以下操作步骤:采用酸浆属一年生草本植物毛酸浆Physalis pubescens L.的干燥茎叶和/或果实为原料,用体积分数为1-99%的乙醇或甲醇的水溶液回流提取1-3次,每次提取1-48小时,合并得到提取液,将提取液去除溶剂得到总浸膏;
将总浸膏用有机溶剂进行萃取,得到有机萃取液,将有机萃取液浓缩后得到有机溶剂层浸膏;所述有机溶剂为甲醇、乙醇、丙酮、乙酸乙酯、氯仿、石油醚及正丁醇中的一种或几种;
将有机溶剂层浸膏通过色谱分离法进行分离,得到上述的化合物;所述提取温度为20-100℃,优选为40-90℃。
本发明第二方面提供了一种包括上述化合物的植物毛酸浆提取物。
本发明第三方面提供了一种药物组合物,包括上述化合物中的一种或几种。
本发明第四方面提供了一种药物组合物,其包括含有治疗有效量的上述化合物或其药学上可接受的盐,和药学上可接受的载体和/或赋形剂。
本发明第五方面提供了上述的化合物、植物毛酸浆提取物及药物组合物在制备抗肿瘤药物中的应用。
所说的肿瘤包括但不限于:各种实体肿瘤和白血病,如肺癌、支气管癌、肝癌、卵巢癌、子宫颈癌、膀胱癌、睾丸癌、肾细胞癌、胰腺癌、胃癌、骨癌、食道癌、结肠癌、胆管癌、前列腺癌、绒毛膜癌、黑色素瘤、胶质细胞瘤、神经纤维瘤、纤维肉瘤、淋巴管瘤等。
含有本发明所述化合物或组合物的治疗泌尿生殖系统癌症的药物可以为适用于口服或注射等应用形式,例如,可为片剂、胶囊剂、粉剂、糖浆剂、针剂等。
本发明优点及效果:(1)提供了一系列结构新颖的化合物及其组合物;(2)运用体外活性筛选体系进行活性评价,发现本发明化合物及其组合物能有效地抑制激素非依赖性前列腺癌细胞、不同种类肾癌细胞以及人恶性黑色素瘤细胞的生长,提示本发明化合物及其组合物可作为治疗肿瘤的药物,具有良好的研究开发前景。
具体实施方式
下面将结合具体实施例对本发明的技术方案进行描述,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1:从毛酸浆茎叶中提取分离活性化合物
茄科酸浆属一年生草本植物毛酸浆Physalis pubescens L.干燥茎叶(9.3kg),用75%乙醇水回流提取2次,每次2小时,合并提取液,减压回收溶剂,浓缩后得到总浸膏(1170.0g)。总浸膏分散到4倍量的水中,分别用等体积的石油醚、乙酸乙酯、正丁醇萃取三次,减压回收溶剂得到石油醚层浸膏15.0g,乙酸乙酯层浸膏99.0g和正丁醇层浸膏200.0g,剩余水层850.0g。
乙酸乙酯层浸膏通过硅胶柱色谱,二氯甲烷-甲醇(100:1~0:100)梯度洗脱,得到9个子馏分(Fr.1~Fr.9)。子馏分Fr.3通过硅胶柱色谱,二氯甲烷-丙酮(100:1~0:100)梯度洗脱,得到8个子馏分(Fr.31~Fr.38),Fr.35经Sephadex LH-20柱色谱,二氯甲烷-甲醇(1:1)洗脱,再经过制备高效液相色谱,甲醇-水(70:30),得到化合物9(8.9mg);Fr.36经Sephadex LH-20柱色谱,二氯甲烷-甲醇(1:1)洗脱,得Fr.361,再经过硅胶柱色谱(环己烷-乙酸乙酯)梯度洗脱,得到2个子馏分(Fr.3611~Fr.3612),Fr.3611经过制备高效液相色谱,甲醇-水(60:40),得到化合物16(17.9mg);Fr.3612经过制备高效液相色谱,甲醇-水(60:40),得到化合物15(85.0mg)和化合物8(19.6mg);子馏分Fr.4通过硅胶柱色谱,环己烷-乙酸乙酯(100:1~0:100)梯度洗脱,得到8个子馏分(Fr.41~Fr.48),Fr.44经SephadexLH-20柱色谱,二氯甲烷-甲醇(1:1)洗脱得子馏分Fr.441,再经过ODS柱色谱,甲醇-水(10:90~100:0)梯度洗脱得子馏分Fr.4413,Fr.4413经过制备高效液相色谱,甲醇-水(70:30),得到化合物10(75.2mg);Fr.46经Sephadex LH-20柱色谱,二氯甲烷-甲醇(1:1)洗脱得子馏分Fr.463,Fr.463过制备高效液相色谱,甲醇-水(60:40),得到化合物11(76.5mg)和化合物17(252.0mg);Fr.47经Sephadex LH-20柱色谱,二氯甲烷-甲醇(1:1)洗脱得子馏分Fr.471,Fr.471过制备高效液相色谱,甲醇-水(50:50),得到化合物12(1600.0mg);Fr.48经Sephadex LH-20柱色谱,二氯甲烷-甲醇(1:1)洗脱得子馏分Fr.481,Fr.481过制备高效液相色谱,甲醇-水(50:50),得到化合物13(132.0mg)。
正丁醇部分(100g),通过硅胶柱色谱,二氯甲烷-甲醇(100:1~0:100)梯度洗脱,共得到9个子馏分(B1~B9)。B2(7.5g)馏分通过硅胶柱色谱,二氯甲烷-甲醇(30:1~0:1)梯度洗脱,得到3个子馏分(B21~B23),B22经过Sephadex LH-20柱色谱,甲醇洗脱得B222,B222经过制备薄层(甲醇为展开剂)、开放ODS柱色谱,甲醇-水(10:90,30:70,40:60,80:20)梯度洗脱和制备高效液相色谱(甲醇:水=50:50)纯化得化合物14(23.0mg);B3(15.2g)馏分通过硅胶柱色谱,二氯甲烷-甲醇(50:1~10:1)梯度洗脱,得到6个子馏分(B31~B36)。子馏分B35经开放ODS柱色谱,甲醇-水(10:90,30:70,50:50,80:20,100:0)梯度洗脱,得8个子馏分(B351~B358),B356中有晶体析出,甲醇重结晶后得化合物2(101.0mg);B6(12g)馏分通过硅胶柱色谱,二氯甲烷-甲醇(6:1)等度洗脱,得到3个子馏分(B61~B63)。B63经中低压ODS柱色谱,甲醇-水(10:90~80:20)梯度洗脱,得14个子馏分(B63-1~B63-14),B63-13经硅胶柱色谱,二氯甲烷-甲醇(5:1)等度洗脱和经Sephadex LH-20柱色谱,甲醇洗脱得B63-13-1,B63-13-1经制备高效液相色谱,甲醇-水(72:28)分离得到化合物4(75.0mg);B7(30g)馏分通过硅胶柱色谱,二氯甲烷-甲醇(8:1~3:1)梯度洗脱,得到4个子馏分(B71~B74),B74馏分经聚酰胺柱色谱,分别以水和乙醇作为洗脱剂得到2个馏分(B741~B742),B742反复经Sephadex LH-20柱色谱,甲醇洗脱,再经制备高效液相色谱,甲醇/水(60:40),分离得到化合物1(43.0mg)。B8(19g)采用反复硅胶柱色谱以二氯甲烷-甲醇系统梯,除去大部分色素后,经反复Sephadex LH-20柱色谱,甲醇洗脱纯化,再通过制备高效液相色谱,甲醇-水(55:45),分离得到化合物3(110.0mg)。
实施例2:从毛酸浆果实中提取分离活性化合物
茄科酸浆属一年生草本植物毛酸浆Physalis pubescens L.的果实(17.5kg),用75%乙醇水回流提取2次,每次2小时,合并提取液,减压回收溶剂,浓缩后得到总浸膏(1420.0g)。总浸膏分散到4倍量的水中,分别用等体积的石油醚、乙酸乙酯、正丁醇萃取三次,减压回收溶剂得到石油醚层浸膏49.0g,乙酸乙酯层浸膏46.0g和正丁醇层浸膏70.0g,剩余水层1246.0g。
乙酸乙酯层浸膏通过硅胶柱色谱,二氯甲烷-甲醇(100:1~0:100)梯度洗脱,得到10个子馏分(E1~E10)。子馏分E3通过硅胶柱色谱,二氯甲烷-甲醇(100:0~0:100)梯度洗脱,得到6个子馏分(E31~E36),E32经Sephadex LH-20柱色谱,二氯甲烷-甲醇(1:1)洗脱,得E322,再经过硅胶柱色谱(环己烷-乙酸乙酯)梯度洗脱,得到8个子馏分(E3221~E3228),E3227经过制备高效液相色谱,甲醇-水(67:33),得到化合物11(92.0mg);子馏分E5通过聚酰胺柱色谱,甲醇-水(10:90~100:0)梯度洗脱,得到3个子馏分(E51~E53),E51通过硅胶柱色谱,(环己烷-丙酮)梯度洗脱,得到12个子馏分(E51-1~E51-12),E51-8通过ODS柱色谱,甲醇-水梯度洗脱,得到6个子馏分(E51-8-1~E51-8-6),E51-8-4经过制备高效液相色谱,甲醇-水(52:48),得到化合物12(19.0mg);E51-10通过ODS柱色谱,甲醇-水梯度洗脱,得到4个子馏分(E51-10-1~E51-10-4),E51-10-3经过硅胶制备薄层色谱(二氯甲烷-甲醇=10:1)得E51-10-3-3,再经过制备高效液相色谱,甲醇-水(48:52),得到化合物18(34.0mg);E52经Sephadex LH-20柱色谱,甲醇洗脱,得5个子馏分(E521~E525),E523经过硅胶制备薄层色谱(环己烷:丙酮=1:1)得E5232,经重结晶(甲醇-水)得化合物5(20.0mg)。子馏分E6通过聚酰胺柱色谱,甲醇-水(10:90~100:0)梯度洗脱,得到6个子馏分(E61~E66),E61经Sephadex LH-20柱色谱,甲醇洗脱,得3个子馏分(E611~E613),E612通过硅胶柱色谱,(二氯甲烷-丙酮)梯度洗脱,得到5个子馏分(E6121~E6125),E6123经过制备高效液相色谱,甲醇-水(67:33),得到化合物13(64.0mg)。子馏分E7经过硅胶制备薄层色谱,二氯甲烷-丙酮(15:1~1:1)梯度洗脱,得到8个子馏分(E71~E78),E74通过ODS柱色谱,甲醇-水梯度洗脱,得到6个子馏分(E741~E746),E746经过制备高效液相色谱,甲醇-水(67:33),得到化合物6(77.0mg)。子馏分E8经Sephadex LH-20柱色谱,甲醇洗脱,得6个子馏分(E81~E86),E83通过ODS柱色谱,甲醇-水梯度洗脱,得到9个子馏分(E831~E839),E839经过制备高效液相色谱,甲醇-水(55:45),得到化合物2(35.0mg)。
正丁醇层浸膏通过硅胶柱色谱,二氯甲烷-甲醇(100:1~0:100)梯度洗脱,得到12个子馏分(B1~B12)。B2通过ODS柱色谱,甲醇-水梯度洗脱,得到8个子馏分(B21~B28),B26通过硅胶柱色谱,二氯甲烷-甲醇(20:1)等度洗脱,得子馏分B261,B261经过制备高效液相色谱,甲醇-水(65:35),得到化合物12(12.2mg)、13(44.2mg)。B11通过硅胶柱色谱,二氯甲烷-甲醇(100:1~0:100)梯度度洗脱,得到6个子馏分(B11-1~B11-6),B11-5通过ODS柱色谱,甲醇-水梯度洗脱,得到14个子馏分(B11-5-1~B11-5-14),B11-5-14经Sephadex LH-20柱色谱,甲醇洗脱,得子馏分B11-5-14-2,B11-5-14-2经过制备高效液相色谱,甲醇-水(70:30),得到化合物4(19.0mg),化合物4经纤维素酶水解得到其苷元,化合物7。B12通过硅胶柱色谱,二氯甲烷-甲醇(100:1~0:100)梯度度洗脱,得到5个子馏分(B12-1~B12-5),B12-5通过ODS柱色谱,甲醇-水梯度洗脱,得到7个子馏分(B12-5-1~B12-5-7),B12-5-4经过制备高效液相色谱,甲醇-水(50:50),得到化合物3(509.0mg);B12-5-5经Sephadex LH-20柱色谱,甲醇洗脱,得子馏分B12-5-5-4,B12-5-5-4经过制备高效液相色谱,甲醇-水(50:50),得到化合物1(24.0mg)。
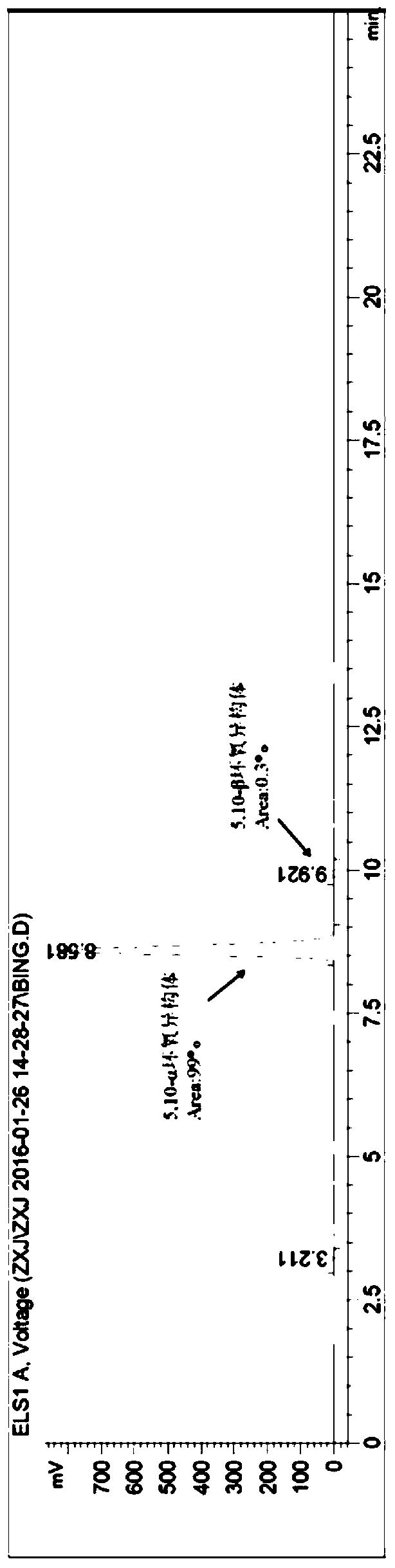
通过理化常数和现代波谱学手段(MS、NMR),结合文献相关数据,鉴定了它们的结构,化合物1-18均为未见文献报道的新化合物,如下所示:
所得各新化合物的物理化学和常数如下:
化合物1无色片晶(甲醇);熔点:241℃; (c 0.06,甲醇);IR(KBr)νmaxcm
化合物2白色粉末; (c=0.07,甲醇);IR(KBr)νmax cm
化合物3白色粉末; (c 0.07,甲醇);IR(KBr)νmax cm
化合物4白色粉末; (c 0.06,甲醇);IR(KBr)νmax cm
化合物5白色粉末; (c=0.07,甲醇);IR(KBr)νmax cm
化合物6白色粉末(甲醇); (c=0.10,甲醇);IR(KBr)νmax cm
化合物7白色粉末(甲醇); (c=0.15,甲醇);IR(KBr)νmax cm
化合物8无色针状结晶(甲醇);熔点:118℃; (c=0.135,甲醇);IR(KBr)νmax cm
化合物9白色粉末; (c=0.10,甲醇);IR(KBr)νmax cm
化合物10白色粉末; (c=0.215,甲醇);IR(KBr)νmax cm
化合物11白色粉末; (c=0.33,甲醇);IR(KBr)νmax cm
化合物12白色粉末; (c=0.545甲醇),IR(KBr)νmax cm
化合物13白色粉末; (c=0.135甲醇);IR(KBr)νmax cm
化合物14白色粉末, (c=0.045甲醇);IR(KBr)νmax cm
化合物15白色针状结晶(甲醇);熔点:195℃; (c=0.145甲醇);IR(KBr)νmax cm
化合物16白色粉末, (c=0.25,甲醇),IR(KBr)νmax cm
化合物17白色粉末; (c=0.345甲醇),IR(KBr)νmax cm
化合物18白色粉末; (c=0.075,甲醇);IR(KBr)νmax cm
实施例3:化合物抗肿瘤活性检测
采用MTT方法,对本发明所公开的化合物进行活性检测,以人前列腺癌细胞(C4-2B和22Rvl),人肾癌细胞(786-0,A-498,Caki-2,ACHN),人黑色素瘤细胞(A375和A375-S2)为例。
取处于对数生长期的上述细胞100μL,每孔2×10
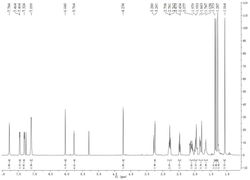
表1化合物1-4的氢谱和碳谱数据
注:上述化合物所用测试溶剂为氘代吡啶;化合物1—4的氢谱测试为600MHz,碳谱测试为150MHz;*表示为一对互变异构体中26S构型异构体的数据。
表2化合物5-8的氢谱和碳谱数据
注:上述化合物5所用测试溶剂为氘代氯仿,化合物6,7所用测试溶剂为氘代吡啶,化合物8所用测试溶剂为氘代甲醇;化合物5、6的氢谱测试为600MHz,化合物7的氢谱测试为400MHz,化合物8的氢谱测试为300MHz,化合物5-7的碳谱测试为150MHz,化合物8的碳谱测试为75MHz;*表示为一对互变异构体中26S构型异构体的数据。
表3化合物9-12的氢谱和碳谱数据
注:上述化合物9-11所用测试溶剂为氘代氯仿,化合物12所用测试溶剂为氘代吡啶;化合物9-12的氢谱测试为600MHz,化合物9的碳谱测试为75MHz,化合物10-12的碳谱测试为150MHz;*表示为一对互变异构体中26S构型异构体的数据。
表4化合物13-16的氢谱和碳谱数据
注:上述化合物13、14所用测试溶剂为氘代吡啶,化合物15、16所用测试溶剂为氘代氯仿;化合物13、14、16的氢谱测试为600MHz,化合物15的碳谱测试为75MHz,化合物14的碳谱测试为100MHz,化合物13、16的碳谱测试为150MHz;*表示为一对互变异构体中26S构型异构体的数据。
表5化合物17-18的氢谱和碳谱数据
注:上述化合物17、18所用测试溶剂为氘代吡啶;化合物17、18的氢谱测试为600MHz,化合物17、18的碳谱测试为150MHz;*表示为一对互变异构体中26S构型异构体的数据。
表6化合物的抗肿瘤活性结果(IC50,μM)
通过表6可以看出,本发明公开的化合物对多种肿瘤细胞增殖具有十分良好的抑制活性,IC50值最小可达0.17μM。通过构效关系分析可知,当本发明所述化合物结构中含有α,β-不饱和酮基团(即R1为羰基,2-3位为双键)和/或5β,6β-环氧基团(即R5与R4结合为基团B),且4位有羟基取代(即R3为羟基)时,其抗肿瘤活性明显优于不含上述基团的化合物。
上述构效关系涉及的有效官能团均属本发明所述化合物特征性结构,同时说明本发明所述化合物是植物毛酸浆发挥药效的有效成分之一。
以上对本发明所提供的抗肿瘤化合物、其提取方法及其应用进行了详细介绍。本文中应用了具体实施例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其中心思想。应当指出,对于本领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护。
一种抗肿瘤化合物、其提取方法及其应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0