









专利摘要
本发明涉及一种配位化合物,其化学式为[VO2(tpy)]ClO4,该化合物具有特效抗癌活性且有望成为低毒高效的抗癌药物,该配位化合物[VO2(tpy)]ClO4的合成包括[VO2(tpy)]ClO4单晶合成及[VO2(tpy)]ClO4微晶或粉末合成,具有独特的抗癌活性,尤其是对肝癌细胞BEL‑7402具有特别强的杀伤力,其IC50=0.4±0.2µM,而传统抗癌药物顺铂在同样的条件下,对BEL‑7402细胞的IC50值为11.5±1.3µM,该配合物的抗肝癌活性是顺铂的近30倍,能达到如此抗癌活性的药物目前还非常少见,这是由配合物独特的结构和钒元素的多氧化态的性质紧密相关的,由于IC50值接近人体所允许的浓度0.3µM,该类化合物并有望开发为新型的低毒、高效的抗癌药物。
权利要求
1.一种配位化合物,其化学式为[VO2(tpy)]ClO4,该配位化合物具有特效抗癌活性且有望成为低毒高效的抗癌药物,其特征是在于,该配位化合物 [VO2(tpy)]ClO4的合成包括[VO2(tpy)]ClO4单晶合成及[VO2(tpy)]ClO4微晶或粉末合成,其中[VO2(tpy)]ClO4单晶合成步骤是:
首先选取VOSO4水溶液与Ba(ClO4)2溶液混合,经搅拌过滤后获得 [VO(H2O)5](ClO4)2水溶液,向该[VO(H2O)5](ClO4)2水溶液加入tpy乙醇溶液获得[VO(H2O)2(tpy)](ClO4)2乙醇、水混合溶液,再通过旋转蒸发除去乙醇获得[VO(H2O)2(tpy)](ClO4)2水溶液,该[VO(H2O)2(tpy)](ClO4)2水溶液则在搅拌的过程下被空气氧化形成[VO2(tpy)]ClO4水溶液,最后经过缓慢蒸发制得[VO2(tpy)]ClO4单晶;
在[VO2(tpy)]ClO4微晶或粉末合成过程中则是将Ba(ClO4)2溶液替换成BaCl2溶液,经搅拌过滤生成[VO(H2O)5]Cl2水溶液,再向[VO(H2O)5]Cl2水溶液加入tpy乙醇溶液获得[VO(H2O)2(tpy)]Cl2乙醇、水混合溶液,再经过旋转蒸发除去乙醇获得[VO(H2O)2(tpy)]Cl2水溶液,该[VO(H2O)2(tpy)]Cl2水溶液再在搅拌的过程下被空气氧化形成[VO2(tpy)]Cl水溶液,向[VO2(tpy)]Cl水溶液中加入饱和NaClO4并不断搅拌最后制备出[VO2(tpy)]ClO4微晶或粉末。
2.如权利要求1所述配位化合物的表征其特征是:包括[VO2(tpy)]ClO4单晶表征及[VO2(tpy)]ClO4微晶或粉末表征,其中[VO2(tpy)]ClO4单晶的表征则通过X-ray单晶衍射获得分子结构键参数及晶体学数据并生成粉末衍射模拟图,[VO2(tpy)]ClO4微晶或粉末表征用X-ray粉末衍射并与单晶的粉末衍射模拟图进行对照,结合电喷雾质谱确定配离子式量及元素分析实验测定C、H、N的含量,从而确定粉末样品的组成,结构与纯度,并通过红外光谱确定官能团结构。
3.一种如权利要求1所述配位化合物的抗癌活性测定方法,其特征在于测定步骤是:首先进行配位化合物[VO2(tpy)]ClO4癌细胞毒实验,测定IC50确定该配位化合物[VO2(tpy)]ClO4最敏感细胞,通过活性氧测定确定配合物是否引发活性氧的产生,通过凋亡实验确定细胞凋亡率,通过线粒体膜电位的测定确定线路提膜的损伤程度,通过彗星实验确定细胞凋亡情况,最后通过上述测定获得凋亡机理。
说明书
技术领域
本发明属于配位化合物技术领域,涉及一种配位化合物的合成、表征与抗癌活性测定方法。
背景技术
钒是生物体中重要的微量元素之一, 在人体中钒的浓度达到0.3μΜ,其化合物已被证实与人类疾病的发病机理和维持正常的生理机能密切相关[1-3]。自1986年Kieler J报道环戊二烯配合物Cp2VCl2在治疗埃利希腹水瘤病中的应用以来[4],含钒化合物的抗癌活性逐渐引起人们的广泛重视。研究表明四价钒的钒氧[VIVO]2+多吡啶配位化合物有望成为高效、广谱、低毒副作用的的抗癌药物[5,6],其中化合物Metvan (双-4,7-二甲基-1,10邻菲啰啉硫酸氧钒)已进入临床试验阶段[7]。然而对五价钒的多吡啶配合物的研究相对较少,且目前所合成的是电中性的分子或含配阴离子的化合物,含配阳离子[VVO2]+的多吡啶配合物还未见报道。
综合以往的研究,本工作的研究意义深远。一方面,与[VIVO]2+的配合物相比,[VVO2]+的配合物作为抗癌药物更具优势:(1) 在溶液和生理条件下,含[VVO2]+的试剂更加稳定,使其在生物体内有足够的时间识别并作用于靶细胞;(2)[VVO2]+具有更为高效的抗癌活性,用药量也会减少,并随之降低毒副作用带来的对患者的身体损害和化疗的风险。这是因为钒的配合物的抗癌作用是通过在机体中产生活性氧(ROS),并由此引发一系列的链式反应和信号传递,最终导致癌细胞凋亡或死亡,产生的活性氧越多,药效越显著。由于[VVO2]+接受电子的能力比[VIVO]2+更强,类似于高价的铬化合物(铬酸盐)在生物体中产生活性氧的作用[8],因此将会在机体内产生更多的ROS。另一方面,与以往合成的五价钒的中性或阴离子的试剂相比,本工作合成的[VVO2(tpy)]+ (tpy = 2,2':6',2''-terpyridine)的优点在于:一价的阳离子配合物,更易穿过细胞膜,甚至是核膜,因此易于被细胞吸收并产生较为丰富的生化反应和优异的活性。这是因为生物体中的阳离子-π的相互作用极为普遍也最为重要,配阳离子与跨膜蛋白质中的具有四偶极子的芳香环的π电子体系产生较强的相互作用,从而能更好地实现跨膜转运[9]。从另一角度来看,tpy是亲脂性的螯合能力强的配体,仅含一分子tpy的配离子体积小且既具有亲水性又具有亲脂性,使其作为药物也易于被生物体吸收与转运。因此,该类化合物作为新型的抗癌药物极具潜力,研究成果有望对癌症的化疗起到不可估量的推动作用。
目前国际上合成含[VVO2]+的化合物,主是腙类化合物作为配体的配位化合物,由于该类配体为负一价的离子L-1,因此所合成的化合物[VVO2L]是中性分子,不能得到配阳离子配合物[10-13]。2000年,Claudia Pifferi等人合成了[VO(tpy)SO4],虽然含有tpy,但由于硫酸根与V有较强的结合,因此只得到了四价钒的三联吡啶配合物[14],未得到与本技术合成的类似的化合物。在我国尚没有出现三联吡啶类钒(V)配合物的合成及生物活性相关的研究论文。
附:参考文献
[1] D. Rehder, The potentiality of vanadium in medicinal applications, Future Med. Chem., 2012, 4, 1823-1837.
[2] D. Rehder, The coordination chemistry of vanadium as related to its biological functions, Coord. Chem. Rev., 1999, 182, 297-322.
[3] M. T. Pepato, N. M. Khalil, M. P. Giocondo, I. L. Brunetti, Vanadium and its complexes: the renewed interest in its biochemistry, Lat. Amer. J. Pharm., 2008, 27,468-76.
[4] P. Köpf-Maier, H. Köpf, Metallocene complexes: organometallic antitumor agents. Drugs Future, 1986, 11, 297-319.
[5] A. Bishayee, A. Waghray, M.A. Patel, M. Chatterjee, Vanadium in the detection, prevention and treatment of cancer: The in vivo evidence, Cancer Letters, 2010, 294,1-12.
[6] M.W. Makinen, M. Salehitazangi, The structural basis of action of vanadyl (VO2+) chelates in cells, Coord. Chem. Rev., 2014, 279, 1-22.
[7] O.J. D’Cruz, F.M. Uckun, Metvan: a novel oxovanadium (IV) complex with broad spectrum anticancer activity, Expert Investig. Drugs, 2002, 11, 1829-1836.
[8] K. Jomova, M. Valko, Advances in metal-induced oxidative stress and human disease, Toxicology, 2011, 283, 65-87.
[9] D.A. Dougherty, Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp, Science, 1996, 271, 163-167.
[10] M.R. Maurya, A.A. Khan, A. Azam, S. Ranjan, N. Mondal, A. Kumar, F. Avecilla, J.C. Pessoa, Vanadium complexes having [VIVO]2+ and [VVO2]+ cores with binucleating dibasic tetradentate ligands: synthesis, characterization, catalytic and antiamoebic activities, Dalton Trans., 2010, 39, 1345-1360.
[11] P.I.S. Maia, F.R. Pavan, C.Q.F. Leite, S.S. Lemos, G.F. Sousa, A.A. Batista, O.R. Nascimento, J. Ellena, E.E. Castellano, E. Niquet, V.M. Deflon, Vanadium complexes with thiosemicarbazones: synthesis, characterization, crystal structures and anti-mycobacterium tuberculosis activity, Polyhedron, 2009, 28, 298-406.
[12] M.R. Maurya, S. Agarwal, M. Abid, A. Azam, C. Bader, M. Ebel, D. Rehder, synthesis, characterization, reactivity and in vitro antiamoebic activity of hydrazone based oxovanadium(IV), oxovanadium(V) and µ-bis(oxo)bis{oxovanadium(V)} complexes, Dalton Trans., 2006, 937-947.
[13] V.M. Deflon, D.M. Oliveira, G.F. Sousa, A.A. Batista, L.R. Dinelli, E.E. Castellano, Oxovanadium(IV,V) complexes with 2-acetylpyridine-2-furanoylhydrazone (Hapf) as ligand. X-ray crystal structures of [VO2(apf)] and [V2O2(µ-O)2(apf)2], Z. Anorg. Allg. Chem., 2002, 628, 1140-1144.
[14] C. Pifferi, M.P. Picchi, R. Cini, Vanadium complexes as models for vanadium species of marine organisms. Synthesis, X-ray structure of oxo(O,O-sulfate)(2,2':6',2''-terpyridine) vanadium (IV) hydrate, and density functional geometry optimization analysis of vanadyl complexes, Polyhedron, 2000, 19, 69-76.
发明内容
为克服上述的技术缺点,本发明提供一种配位化合物的合成、表征与抗癌活性测定方法,它能够避免使用阴离子配体和配位能力强的阴离子作为抗衡离子,通过控制反应条件,使四价的钒配合物充分氧化,具有独特的抗癌活性,尤其是对肝癌细胞BEL-7402具有特别强的杀伤力。
本发明解决其技术问题所采用的技术方法是:一种配位化合物,其化学式为 [VO2(tpy)]ClO4,该配位化合物具有特效抗癌活性且有望成为低毒高效的抗癌药物,该配位化合物 [VO2(tpy)]ClO4的合成包括[VO2(tpy)]ClO4单晶合成及[VO2(tpy)]ClO4微晶或粉末合成,其中[VO2(tpy)]ClO4单晶合成步骤是:
首先选取VOSO4水溶液与Ba(ClO4)2溶液混合,经搅拌过滤后获得 [VO(H2O)5](ClO4)2水溶液,向该[VO(H2O)5](ClO4)2水溶液加入tpy乙醇溶液获得[VO(H2O)2(tpy)](ClO4)2乙醇、水混合溶液,再通过旋转蒸发除去乙醇获得[VO(H2O)2(tpy)](ClO4)2水溶液,该[VO(H2O)2(tpy)](ClO4)2水溶液则在搅拌的过程下被空气氧化形成[VO2(tpy)]ClO4水溶液,最后经过缓慢蒸发制得[VO2(tpy)]ClO4单晶;
在[VO2(tpy)]ClO4微晶或粉末合成过程中则是将Ba(ClO4)2溶液替换成BaCl2溶液,经搅拌过滤生成[VO(H2O)5]Cl2水溶液,再向[VO(H2O)5]Cl2水溶液加入tpy乙醇溶液获得[VO(H2O)2(tpy)]Cl2乙醇、水混合溶液,再经过旋转蒸发除去乙醇获得[VO(H2O)2(tpy)]Cl2水溶液,该[VO(H2O)2(tpy)]Cl2水溶液再在搅拌的过程下被空气氧化形成[VO2(tpy)]Cl水溶液,向[VO2(tpy)]Cl水溶液中加入饱和NaClO4并不断搅拌最后制备出[VO2(tpy)]ClO4微晶或粉末。
配位化合物的表征包括[VO2(tpy)]ClO4单晶表征及[VO2(tpy)]ClO4微晶或粉末表征,其中[VO2(tpy)]ClO4单晶的表征则通过X-ray单晶衍射获得分子结构键参数及晶体学数据并生成粉末衍射模拟图,[VO2(tpy)]ClO4微晶或粉末表征用X-ray粉末衍射并与单晶的粉末衍射模拟图进行对照,结合电喷雾质谱确定配离子式量及元素分析实验测定C、H、N的含量,从而确定粉末样品的组成,结构与纯度,并通过红外光谱确定官能团结构。
该配位化合物的抗癌活性测定方法,其测定步骤是:首先进行配位化合物[VO2(tpy)]ClO4癌细胞毒实验,测定IC50确定该配位化合物[VO2(tpy)]ClO4最敏感细胞,通过活性氧测定确定配合物是否引发活性氧的产生,通过凋亡实验确定细胞凋亡率,通过线粒体膜电位的测定确定线路提膜的损伤程度,通过彗星实验确定细胞凋亡情况,最后通过上述测定获得凋亡机理。
本发明的有益效果是:首先利用中性的三联吡啶配体,采用弱配位作用的抗衡阴离子及其充分氧化的条件下合成了固相以及溶液相在近生理条件下(pH约为7)均稳定的含五价钒三联吡啶配阳离子的配合物及其单晶,该类配合物在国内、国际均未见报道;其次合成的配合物具有独特的抗癌活性,尤其是对肝癌细胞BEL-7402具有特别强的杀伤力,其IC50= 0.4 ± 0.2 µM,而传统抗癌药物顺铂在同样的条件下,对BEL-7402细胞的IC50值为11.5 ± 1.3 µM,因此本发明合成的配合物的抗肝癌活性是顺铂的近30倍,能达到如此抗癌活性的药物目前还非常少见,这是由配合物独特的结构和钒元素的多氧化态的性质紧密相关的,由于IC50值接近人体所允许的浓度0.3 µM,该类化合物并有望开发为新型的低毒、高效的抗癌药物。
附图说明
图1是合成配合物[VO2(tpy)]ClO4单晶方框结构示意图;
图2是合成配合物[VO2(tpy)]ClO4(微晶或粉末)方框结构示意图;
图3是配合物[VO2(tpy)]ClO4单晶及[VO2(tpy)]ClO4微晶或粉末表征示意图;
图4是配合物[VO2(tpy)]ClO4抗癌活性的测定方案示意图;
图5是单晶配合物的X-ray衍射测定的单元结构图;
图6是细胞BEL-7402的线粒体膜电位测定实验(a)空白组,(b)(c)分别用0.5 µM和1.0 µM的对照图;
图7是配合物[VO2(tpy)]ClO4的晶体数据和结构精修参数图;
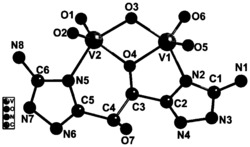
图8是配阳离子的键长和键角示意图;
图9是粉末衍射图谱;
图10是配合物对所选细胞株IC50值示意图;
图11是细胞BEL-7402的EB染色彗星实验(a)EB染色的控制组,(b)加入0.5 µM的配合物孵育24小时,b中的细胞呈现明显慧尾对照图。
具体实施方式
下面结合附图及实施例对本发明进一步说明。
实施例1:
配合物[VO2(tpy)]ClO4单晶制备和单晶结构的测定
参见图1,制备过程是:首先取0.6 mmol VOSO4∙3H2O溶于25 ml水中得溶液A,称取0.6 mmol Ba(ClO4)2溶于25 ml水中得溶液B,搅拌下将溶液B缓慢滴加到溶液A中,产生BaSO4白色沉淀,过滤得蓝色溶液C。将含0.5 mmol 的tpy的乙醇溶液(15 ml),在搅拌下滴加到溶液C中,室温搅拌30分钟,得一绿色溶液,旋转蒸发至约25 ml左右,室温搅拌5天,至溶液变成橙黄色,过滤,滤液在室温下缓慢自由蒸发,45天左右得橙色块状晶体[VO2(tpy)]ClO4。
参见图3,单晶结构测定:将配合物单晶粘在玻璃毛上,插入铜管后,将其置于Bruker SMART 1000 CCD四圆衍射仪的样品台上,用石墨单色器分光的Mo-Kα(λ= 0.71073 Å)射线作为光源,在296K下进行测定。采用ω扫描技术在1.78 < θ< 25.00° 范围内收集衍射强度数据,收集4009个衍射点。晶体结构用SHELXTL-97的直接法解出,采用全矩阵最小二乘法在F2上进行个向异性修正。最后对2745个观测点 (F ≥ 4.0 σ(F))及235个变量的精修获得收敛。在对所有的非氢原子用各向异性位移参数精修之后,对配体上的氢原子的坐标及其位移参数进行各向同性的精修。在精修中,氢原子将被相对固定在其母原子上,随母原子的移动而移动,即骑在母原子上,氢原子和碳原子的距离利用程序的预设值C—H = 0.96 Å。所有氢原子都进行各向同性温度因子校正。有关的晶体学数据和结构精修参数见图7。配合物阳离子的部分键长,键角列于图8,参见图5。
实施例2:
配合物[VO2(tpy)]ClO4微晶制备及表征
参见图2,制备:取0.6 mmol VOSO4∙3H2O溶于25 ml水中得溶液A,称取0.6 mmol BaCl2溶于25 ml水中得溶液B,搅拌下将溶液B缓慢滴加到溶液A中,产生BaSO4白色沉淀,过滤得蓝色溶液C。将含0.5 mmol 的tpy的乙醇溶液(15 ml),在搅拌下滴加到溶液C中,室温搅拌30分钟,得一绿色溶液,旋转蒸发除去乙醇,加水稀释至溶液的总体积80 ml,室温搅拌7天,至溶液变成橙黄色,过滤,于搅拌条件下,在滤液中滴加饱和NaClO4溶液,至沉淀完全,继续室温搅拌4小时,得橙黄微晶,抽滤,用水洗涤3次(每次5 ml),再用乙醇洗涤两次(每次5 ml),放入真空干燥器中3天得产品,收率42%。
参见图3,表征:
(1) 粉末衍射:数据和实验图谱在Bruker D8 Advance X-ray衍射仪上进行测定和获取,其中Cu-Kα靶,λ=0.154 nm,石墨单色器衍射束单色比,高压50 KV,管流20 mA。模拟衍射图谱根据X-ray单晶数据,采用SHELXTL-XPOW程序计算获得。通过实验图谱(Experimental)和模拟图谱(Simulated),见图9,峰的位置和相对强度高度一致,这说明所得微晶具有单晶同样的结构;
(2) 红外光谱:利用Bruker VECTOR22 FT-IR红外光谱仪和KBr压片技术进行测定,各峰指派为 (cm-1):1600, 1480 (C=Ctpy, C=Ntpy), 1080, 953 (V=O);
(3) 电喷雾质谱:电喷雾质谱(ES-MS)用LCQ系统 (Finnigan MAT, USA) 记录,选用DMSO作流动相。[DMSO, m/z]: 316.6 ([VO2(tpy)]+), 实验值与理论值吻合。
(4)元素分析:元素分析(C、H、N)用Elementar Vario EL元素分析仪测定。测试结果(%): C 43.37, H 2.81, N 10.05。计算值 (C15 H11 ClN3 O6 V, %): C 43.34, H 2.67, N 10.11。实验值与计算值基本一致。
参见图4,配合物[VO2(tpy)]ClO4抗肿瘤活性研究
(1) 体外细胞毒性实验:收集对数生长期的细胞,调整细胞悬液浓度为5 × 104 ~ 1 .5 ×105个/m L,取96孔板每孔100 μL。将接种好的96孔板置于培养箱中孵育(5% CO2,37oC),待孔底单层细胞长至80%左右时弃掉培养液,加入1640 培养液90μL和设定好浓度梯度的药物10μL,继续孵育 48 h,弃去培养液,补加90 μL1640 培养液和10 μL的MTT (5 mg∙m L-1),培养箱中继续孵育4 h。取出至培养箱外,弃去培养液,每孔加入100 μL的 DMSO,振摇10 min,用酶标仪测定其490 nm处的吸光度值,计算IC50值结果见图10,结果显示,本合成的化合物,比目前广泛使用的抗癌药物顺铂(cisplatin)的活性都好,尤其是对肝癌细胞BEL-7402,本化合物的抗癌活性是顺铂的近30倍。以下实验(2)至(5)均采用BEL-7402细胞。
(2) AO/EB 染色检测凋亡实验:取12孔板,每孔接种细胞1×105 ~ 2×105个。待单层细胞长至90%左右时,加入相应浓度的药物。培养箱中孵育24 h后,弃去培养液,PBS洗两遍。滴加AO/EB染色液(100 μg∙m L-1)覆盖玻片,37oC染色30 min,弃去染液,PBS洗涤三次,荧光显微镜下观察、拍照记录,仅0.5 µM的配合物作用BEL-7402细胞24 h,然后对细胞进行AO/EB双染,细胞被AO染色成绿色,凋亡细胞染色质固缩染色加深;但EB没能透过细胞膜 (未出现呈橘红色),这正是细胞早期凋亡的特征。说明这个配合物能高效率地诱导肝癌细胞BEL-7402的凋亡。
(3) 彗星实验:即单细胞凝胶电泳实验,BEL-7402细胞在0.5 µM的配合物作用下,37oC孵育24 h,并用胰蛋白酶化法收集细胞。三层法制备凝胶板,电解液为300 mM NaOH, 1.2 mM EDTA,电泳在25 V,300 mA下进行。电泳后用400 mM Tris, HCl, pH 7.5的溶液将胶板洗至中性,用EB (20μg/m L)在暗处染色20分钟,用荧光显微镜拍照。参见图11,和对照组比较,经配合物处理的细胞均出现慧尾,这说明配合物进入到细胞核,与DNA作用,并使DNA断裂不同长度的碎片,这是细胞凋亡重要的启动信号。
(4) 线粒体膜电位检测:收集对数期细胞,调整细胞悬液浓度。取12孔板,每孔接种1× 105 ~ 2 × 105个。待单层细胞长至80%左右时,加入相应浓度的药物。培养箱中孵育24 h后,往阳性对照组加入1 μL的CCCP继续孵育20 min,弃去12孔板中的培养液,用冷的PBS洗涤三次,加入0.5 m L细胞培养液和0.5 m L JC-1染色工作液充分混匀。将细胞置于培养箱中37oC孵育20 min,与此同时取3 m L JC-1染色缓冲液加入12 mL双蒸水稀释5倍置于冰上。待孵育结束后,弃掉染色液,并用JC-1染色缓冲液洗涤两次,加入1 m L细胞培养液,在荧光显微镜下观察,拍照记录。线粒体膜电位的检测以JC-1 为荧光探针,细胞内线粒体膜电位较高时JC-1以聚合物的形式存在于线粒体基质中发红色荧光,细胞内线粒体膜电位较低时JC-1以单体的形式存在发绿色荧光,所以检测 JC-1 的荧光就可以检测到线粒体膜电位的变化。细胞凋亡早期细胞线粒体内膜通透性会增加,线粒体膜电位会发生下降,一旦线粒体膜电位崩溃细胞就只能走向凋亡。本方法合成的配合物作用BEL-7402 细胞后,JC-1染色细胞,荧光显微镜拍照记录,可见线粒体膜电位检测实验结果显示配合物 能使 BEL-7402 细胞线粒体膜电位降低。参见图6:
该图为利用JC-1染色法分析细胞BEL-7402的线粒体膜电位测定实验(a)空白组,(b)(c)分别用0.5 µM和1.0 µM的配合物孵育24小时,b, c组均全部成为绿色荧光,说明配合物的作用使线粒体膜电位下降,这是线粒体释放细胞色素,并引发细胞凋亡的链式反应的关键。
(4) 活性氧检测:收集对数期细胞,调整细胞悬液浓度。取6孔板,每孔接种4 × 105个。待单层细胞长至90%左右时,加入相应浓度的药物,培养箱中孵育24 h后,弃去6孔板中的培养液,用PBS洗涤两次,1 m L胰酶消化,收集细胞于2 m L的EP管中离心(3000 rpm,5 min),弃掉上清液,加入1 m L不含血清的培养液重悬细胞,离心(3000 rpm,5 min),弃掉上清液,加入1︰1000稀释好的DCFH-DA,置于细胞培养箱中37oC孵育20 min,每隔3~5 min颠倒混匀一次,然后用不含血清的培养液洗涤两次,离心(3000 rpm,5 min),弃掉上清液并加入300 μL无血清的培养基混匀并移入流式管,上机检测。配合物[VO2(tpy)]ClO4作用BEL-7402细胞后二氯二氢荧光素-乙酰乙酸酯(DCFH-DA)染色细胞,流式细胞仪检测DCF荧光强度, DCFH-DA本身没有荧光,能穿过细胞膜进入细胞,进入细胞后被细胞内的酯酶水解成DCFH,而DCFH不能穿过细胞膜,DCFH进一步被细胞内的活性氧氧化成发绿色荧光的DCF,所以可以通过DCF的荧光强度来反映细胞内的活性氧水平。实验结果表明配合物能升高BEL-7402细胞内的活性氧水平。参见图7:
该图为利用H2DCF-DA作为荧光探针对BEL-7402细胞内活性氧的测定实验(a)空白组,(b)Rosup阳性控制组,(c)用0.5 µM的配合物孵育24小时。 c组与b一样,发出绿色的荧光,这说明进入细胞的配合物能诱导了活性氧的产生,因而产生的活性氧使DCFH发生氧化生成发荧光的DCF。
综合以上抗癌活性实验,初步推测抗癌机理为:配合物进入细胞核,产生活性氧,氧化断裂DNA,产生凋亡信号,并移位至线粒体,使线粒体膜电位崩溃,进一步释放释放凋亡信号,如细胞色素C,并引发一系列的级联反应(如caspase级联反应),导致癌细胞凋亡,有关详细而深入的抗癌机理的有待于进一步系统的研究。
结论:本方法的重复性好,产品纯度高。所合成的[VO2(tpy)]ClO4具有极高的抗癌活性,有望开发为新型的高效、广谱、低毒副作用的癌症治疗的化疗药物。
一种配位化合物的合成、表征与抗癌活性测定方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0