








专利摘要
本发明公开了一种磁珠的制备方法及应用,属于磁性材料制备技术领域。该制备方法为,首先制备表面功能化的磁性纳米粒子,然后采用交联剂将至少两个表面功能化的磁性纳米粒子连接起来,制备出磁含量为70%‑98%的磁珠。本发明解决了现有磁珠磁含量低以及为了实现快速分离,极大地增加磁珠体积而导致悬浮稳定性降低的问题。另外,对于被捕获组分生物反应特别是免疫反应来说,本发明制得的小体积磁珠所导致的空间位阻显著降低,捕获能力和分析检测灵敏度提高,能够更好地用于生物医药分离与分析,特别适用于生物体液或反应液中细胞、细菌、病毒、DNA/RNA、蛋白质等的分离以及基于化学发光、电化学发光和荧光的临床生化快速自动化检测。
权利要求
1.一种磁珠的制备方法,其特征在于:首先制备表面功能化的磁性纳米粒子,然后采用交联剂将至少两个表面功能化的磁性纳米粒子连接起来,制备出磁含量为70%-98%的磁珠;所述表面功能化的磁性纳米粒子为柠檬酸修饰的磁性纳米粒子;所述交联剂为Mw=1800的聚醚酰亚胺。
2.根据权利要求1所述的磁珠的制备方法,其特征在于:所述磁珠的粒径至少为所述磁性纳米粒子粒径的2倍,具体为200nm-50μm。
3.根据权利要求1所述的磁珠的制备方法,其特征在于:所述磁珠为球形或不规则形。
4.根据权利要求1所述的磁珠的制备方法,其特征在于:所述磁珠在溶液中的完全分离时间为1min-6min。
5.一种如权利要求1-4任一项所述的磁珠的制备方法制得的磁珠在生物医药分析与分离的应用,其特征在于:将所述磁珠直接加入生物样品中,利用电荷相互作用进行目标物的磁性快速捕获分离与分析;
或者首先在所述磁珠上偶联捕获试剂,再进行目标物的磁性快速捕获分离与分析。
6.根据权利要求5所述的磁珠在生物医药分析与分离的应用,其特征在于:所述捕获试剂为抗体或适体。
说明书
技术领域
本发明属于磁性材料制备技术领域,尤其涉及一种磁珠的制备方法及应用。
背景技术
对于常规方法合成的磁性纳米粒子,通常表现出以下两方面的显著特征。第一,表现出超顺磁性或近似超顺磁性(剩磁较少或极少),有利于在磁场作用下从分散的溶液中聚积而分离,同时也有利于取消磁场作用后再次分散到溶液中,而实现对分离组分的分析,上述特征特别有利于自动化分离与分析;第二,粒径(指用TEM表征的非水合粒径)较小,通常在10-100nm,每个粒子在强磁场作用下,表现出较小的磁力,从分散的溶液中分离时间长、分离不完全,从而导致分析速度慢、分析灵敏度降低。
为了利用上述优点(超顺磁性或近似超顺磁性)和克服上述缺点(分离时间长、分离不完全),必须将多个磁性纳米粒子组装成为一个粒径更大的磁珠(为了下述表达的方便,磁性纳米粒子即指未连接的、小粒径的、纳米尺度的粒子,磁珠即指连接后的、大粒径的粒子)。一个磁珠的磁力即由多个磁性纳米粒的磁力加和,使磁珠的磁力远大于磁性纳米粒子,在磁场作用下能够快速集聚从而在分散的溶液中快速、完全分离。同时,由于磁珠是由磁性纳米粒子组装而成,即保留了磁性纳米粒子的超顺磁性或近似超顺磁性,在取消磁场作用后即可实现磁珠的再次快速分散。对于生物分离与分析使用的磁珠,其实还有另一个重要的要求,在溶液中必需具有良好的悬浮稳定性,否则不能与被分离组分在一定的反应时间内、且在悬浮状态下进行快速、完全反应。因此,对于一个性能卓越的生物分离与分析使用的磁珠必须具备三方面的条件,其一,强磁力,最好在1-2分钟内在常用的强磁铁(钕铁硼磁铁)作用下,实现分散在溶液中的磁珠的快速、完全分离;其二,超顺磁性或近似超顺磁性,使磁珠在较弱外力作用下(如振摇、液体流动)再次快速分散;其三,良好的悬浮稳定性,在水溶液中至少30分钟内能够悬浮稳定,不发生沉降。
对于现行的磁珠,通常在强磁力和悬浮稳定性间存在矛盾。为了获得强磁力,必须提高磁珠中磁性纳米粒子的数量,但是磁珠的体积往往会快速增大,导致悬浮稳定性降低,同时也导致磁珠对捕获对象的空间位阻增大,捕获能力降低。磁珠体积快速增大的主要因素,一方面是磁性纳米粒的积聚,另一方面是用于固载磁性纳米粒的非磁性组分的化学聚积。前者是不能回避的,因此,解决磁珠快速增大的唯一办法即是降低后者在磁珠中的含量,即提高磁珠的磁含量(磁含量即为磁珠中磁性物质(磁性纳米粒)的含量)。现行的商品化磁珠,包括医用磁珠,大多数均是高分子聚合物磁珠,包括均聚物和共聚物磁珠。常用的合成高分子包括聚苯乙烯、聚丙烯酸、聚甲基丙烯酸、聚乙二醇、聚乙烯比咯烷酮、聚乳酸、聚乙烯醇以及他们的共聚物等。常用的制备方法包括包埋法、分散聚合法、乳液聚合法和活性聚合法等。这些方法均是在有机聚合单体发生聚合反应的过程中,将磁性纳米粒包覆在高分子微球之中,从而使一个高分子磁珠中固载多个磁性纳米粒。为了让磁性纳米粒稳定地固载,必须要求磁珠中的每一个磁性纳米粒均要有足够的高分子包覆层,这样即导致了磁珠中非磁性物质含量的增加,即磁含量较低。除了高分子磁珠外,事实上,还有无机材料磁珠。采用水解有机硅氧烷的方法制备SiO2包覆的磁珠,制备方法主要包括反相微乳法、气溶胶高温分解法和溶胶凝胶法等。在保证磁力的前提下,这类磁珠的磁含量更低。上述两类磁珠,均是在制备非磁性微球的过程中通过包覆(包括物理包覆和基于化学键固载的包覆)固载磁性纳米粒,这是不可避免地增加非磁性物质的含量,从而降低制备磁珠的磁含量,这是一个内在的固有矛盾,难于从方法上避免。
发明内容
本发明的目的是提出一种磁珠的制备方法,该制备方法利用交联剂的活泼基团和磁性纳米粒子的表面功能基团之间的化学反应所形成的化学键,将多个磁性纳米粒子连接起来,制备出高磁含量的磁珠。
为达此目的,本发明采用以下技术方案:
发明提供了一种磁珠的制备方法,首先制备表面功能化的磁性纳米粒子,然后采用交联剂将至少两个表面功能化的磁性纳米粒子连接起来,制备出磁含量为70%-98%的磁珠。
作为一种优选方案,所述表面功能化的磁性纳米粒子的粒径为5-250nm,所述表面功能化的磁性纳米粒子为表面修饰的磁性纳米粒子或包埋大分子的磁性纳米粒子或表面固载大分子的磁性纳米粒子。
作为一种优选方案,所述表面功能化的磁性纳米粒子的表面功能基团为羟基或羧基或氨基或醛基或巯基中的一种或几种。
作为一种优选方案,所述交联剂采用含有活泼基团的大分子物质,所述活泼基团为羟基或羧基或醛基或巯基或氨基中的一种或几种。
作为一种优选方案,所述大分子物质为聚合物或嵌段聚合物或蛋白质或DNA或多肽或多糖中的一种或几种。
作为一种优选方案,所述磁珠的粒径至少为所述磁性纳米粒子粒径的2倍,具体为200nm-50μm。
作为一种优选方案,所述磁珠为球形或不规则形。
作为一种优选方案,所述磁珠在溶液中的完全分离时间为1min-6min。
本发明还提供了一种如上任一所述的磁珠的制备方法制得的磁珠在生物医药分析与分离的应用,将所述磁珠直接加入生物样品中,利用电荷相互作用进行目标物的磁性快速捕获分离与分析;
或者首先在所述磁珠上偶联捕获试剂,再进行目标物的磁性快速捕获分离与分析。
作为一种优选方案,所述捕获试剂为抗体或适体。
本发明的有益效果为:
本发明提供了一种磁珠的制备方法,本发明利用交联剂的活泼基团和磁性纳米粒子的表面功能基团之间的化学反应所形成的化学键,将多个磁性纳米粒子连接起来,制备出高磁含量(70%-98%)的磁珠。本发明制备方法简单,制备产率高,磁珠粒径大小可控;在制备磁珠中固载相同量的磁性纳米粒,即每个磁珠具有相同磁力的前提下,本发明制备的磁珠的磁含量更高,体积更小,表面电荷丰富,表面亲水性更强,悬浮稳定性更好,解决了现有磁珠特别是医用磁珠磁含量低的问题,也克服了现有磁珠为了实现快速分离,极大地增加磁珠体积而导致悬浮稳定性降低的问题。
本发明还提供了一种上述的磁珠的制备方法制得的磁珠在生物医药分析与分离的应用,将所述磁珠直接加入生物样品中,利用电荷相互作用进行目标物的磁性快速捕获分离与分析;或者首先在所述磁珠上偶联捕获试剂,再进行目标物的磁性快速捕获分离与分析。对于被捕获组分生物反应特别是免疫反应来说,本发明制得的小体积磁珠所导致的空间位阻显著降低,捕获能力和分析检测灵敏度提高,能够更好地用于生物医药分离与分析,特别适用于生物体液或反应液中细胞、细菌、病毒、DNA/RNA、蛋白质等的分离以及基于化学发光、电化学发光和荧光的临床生化快速自动化检测。
附图说明
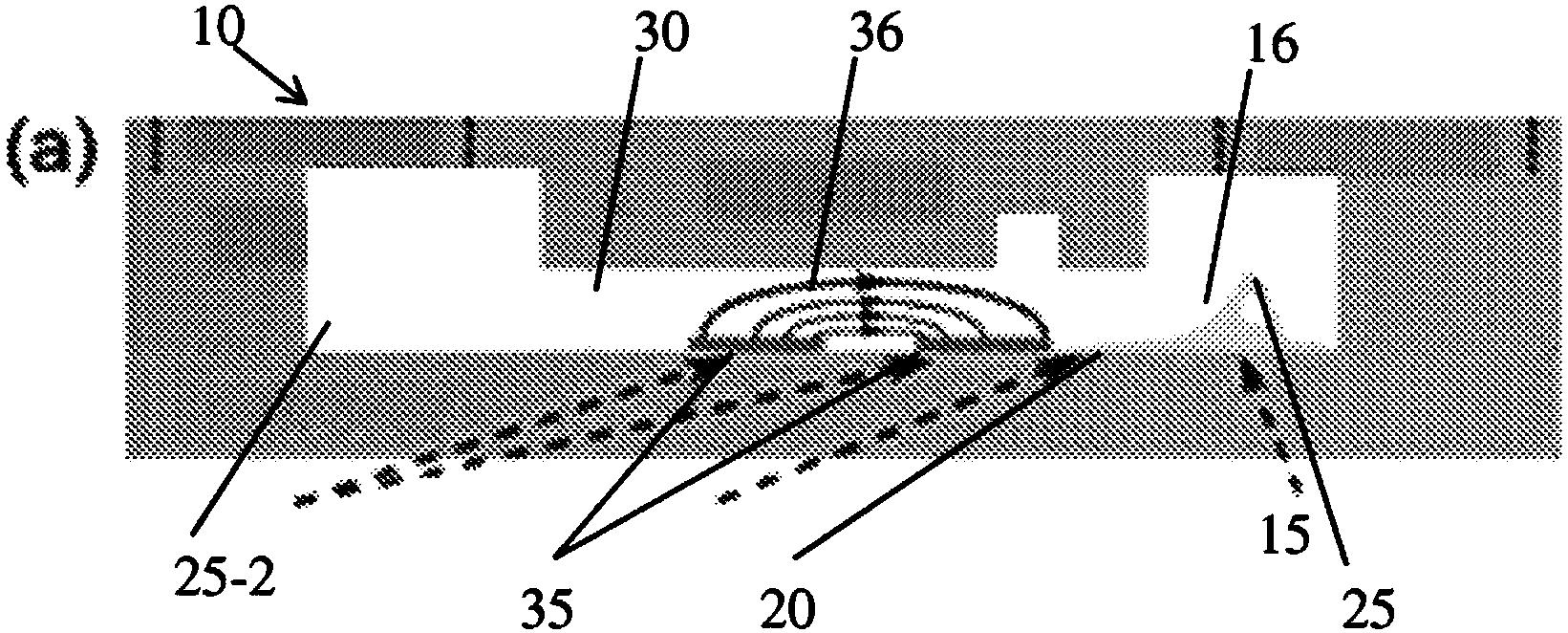
图1是本发明实施例一的PEI交联磁珠的热失重曲线图;
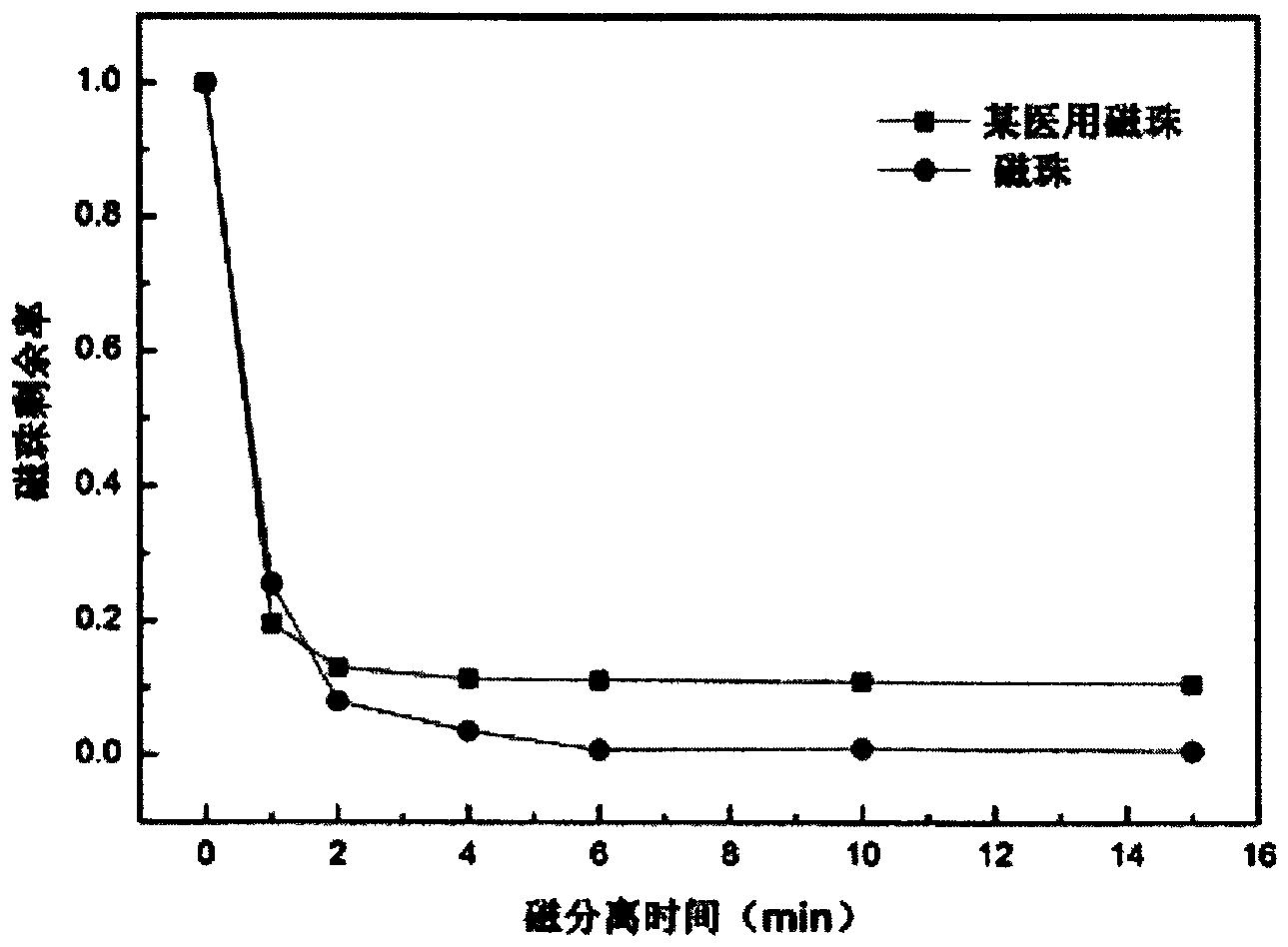
图2是本发明实施例一的PEI交联磁珠与某流行医用磁珠比较,在磁分离时磁珠的剩余率(溶液中未被分离磁珠的百分率)随时间的变化曲线图;
图3是本发明实施例一的PEI交联磁珠溶液的紫外吸收光谱的模随时间的变化曲线图。
具体实施方式
下面结合实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
为使本发明解决的技术问题、采用的技术方案和达到的技术效果更加清楚,下面将结合附图对本发明实施例的技术方案作进一步的详细描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
众所周知,表面功能化的磁性纳米粒子主要分为以下几种:
1、未表面修饰的磁性纳米粒子:用常规方法制备的磁性纳米粒子,包括Fe3O4、Fe2O3及其它们的杂合体、CoFe2O4、NiFeO4、ZnFe2O4、MnFeO4等,表面的众多功能基团是指大量的羟基(-OH)。
2、表面修饰的磁性纳米粒子:对于用常规方法制备的磁性纳米粒子,通过化学修饰改变粒子表面的功能基团,相应的功能基团可以是羟基或羧基或氨基或醛基或巯基的其中一种或几种。
3、包埋大分子的磁性纳米粒子:用常规方法制备磁性纳米粒子的同时,加入需要包埋的大分子,使其大分子部分被包埋在粒子内部,另一部分在粒子外部。但不排除有些大分子是被吸附在粒子的表面。粒子外部的大分子的活泼基团即为表面功能基团。
4、表面固载大分子的磁性纳米粒子:用常规方法制备磁性纳米粒子之后,利用化学键、疏水作用、电荷作用中的一种或几种作用力将大分子固载于粒子表面。但不排除有些大分子是被吸附在粒子的表面。粒子外部的大分子的活泼基团即为表面功能基团。
实施例一
本实施例提供了一种磁珠的制备方法,包括以下步骤:
(1)柠檬酸修饰的磁性纳米粒子制备:将制备的Fe3O4磁性纳米粒子水基分散液超声分散15min,取20mL置于三颈瓶内通氮气除氧20min,加入到一定量的柠檬酸钠溶液中超声15min,使Fe3O4磁性纳米粒子在体系中分散均匀,连接实验装置,保持氮气环境,将机械搅拌的速度调至600rpb,水浴锅温度升至60℃后搅拌反应1h,用钕铁硼磁铁进行磁分离,三蒸水洗涤去除多余的柠檬酸钠,直至磁分离不能获得澄清的上清液为止,最后重新分散于20mL三蒸水中,制得柠檬酸修饰的水溶性Fe3O4,即柠檬酸修饰的磁性纳米粒子,表面功能基团即为羧基。
(2)磁性纳米粒子的PEI交联制备磁珠:在15mg磁性纳米粒子的水基分散液中,采用EDC·HCl和sulfo-NHS活化羧基,37℃的反应条件下摇床活化1h,磁分离,水洗三次,将其加入圆底烧瓶中在37℃水浴条件下超声分散1min;取12.5mg的聚合物PEI(Mw=1800)水溶液于圆底烧瓶中,并加入pH=7.4的缓冲溶液后置于摇床中振摇,反应1h,磁分离,水洗,复溶于水中,即得PEI交联的磁珠。
本实施例制备的磁珠的粒径和Zeta电位分析:
从附表1可以看出,对于动态光散射法测定的粒径,本实施例制备的磁珠的ChiSquared值和P.I.值均较小,说明粒径分布很好地满足于正态分布,且分布均匀。并且,磁珠带有丰富的表面电荷,其相应的粒径远小于现有医用磁珠的粒径(约2700nm)。上述特征使其在水溶液中表现出了良好的悬浮稳定性。更为重要的是,小尺度磁珠将极大地减小磁珠对被捕获组分的空间位阻,增大捕获能力,提高检测灵敏度,减少捕获时间,实现快速分析检测。
表1Mw=1800的PEI制备的磁珠的粒径和Zeta电位
本实施例制备的磁珠的热失重曲线和磁含量分析:
我们对PEI交联(Mw=1800)制备的磁珠做了热失重表征,如附图1所示,从热重曲线上我们可以看出,失重是分两个阶段的:第一阶段是温度在140℃以下时,这段的失重是磁珠表层中的水蒸发,此时磁珠磁含量为99.12%;第二阶段是从200℃开始的,这是由于磁珠中所含的非磁性组分(包括表面功能化的柠檬酸和大分子PEI)分解造成的失重。温度在640℃以后,热失重曲线趋于稳定,为剩余的Fe3O4。以包括水份在内的非磁性组分的热失重率为基础,计算磁珠的磁含量为91.87%。当然,除去水份的影响,磁含量会更高,这极其有利于强磁力、小体积、高悬浮稳定性的高效磁珠制备。
本实施例制备的磁珠与医用磁珠的磁分离性比较:
在磁分离时,以溶液中未被分离磁珠占总磁珠的百分率(本发明定义为“磁珠剩余率”)为标准,考察以PEI交联磁珠与某流行医用磁珠的磁分离能力。从附图2可见,在外加磁场作用1min时,未被分离的剩余率,医用磁珠略低于本实施例的PEI交联磁珠,但在2min后PEI交联磁珠的剩余率便小于医用磁珠的剩余率。PEI交联磁珠在6min内实现了近乎100%分离,但是,医用磁珠长时间均不能实现100%分离,其剩余量高达10%以上,这就预示着有高达10%以上的被分离组分不能实现分离。对于临床检验中的高灵敏的发光分析(如广泛使用的化学发光、电化学发光分析)而言,预示着大量的低含量样本将出现假阴性。这对于现在临床广泛使用的癌症早期诊断的生物标记物检测,是极其不利的,极易出现大量漏检。由此表明,本实施例制备的磁珠,磁分离性远优于某流行医用磁珠。
本实施例制备的磁珠和医用磁珠的悬浮稳定性比较:
由于磁珠的紫外光谱没有特征吸收,以紫外吸光光谱的模随时间的变化曲线来表征溶液中的磁珠含量。仍以某流行医用磁珠为比较标准,从附图3可见,在20min内两类磁珠在溶液中的悬浮稳定性不存在显著差异,均表现出了良好的稳定性。但是,随着时间的增加,两者的差异就极其明显,在长达120nm时,本实施例制备的磁珠仍然是极其稳定的,没有发现明显的沉降,但对照的医用磁珠却沉降了15%。表明本实施例制备的磁珠具有极好的悬浮稳定性,远优于某流行医用磁珠。
实施例二:
本实施例提供的一种磁珠的制备方法与实施例一所述的磁珠的制备方法基本相同,区别之处在于:
(1)具有小分子功能基团的表面修饰的磁性纳米粒子的制备:将含有功能基团(包括羟基、氨基、醛基、巯基和不同于柠檬酸的其它羧酸所含有的羧基)的小分子功能化试剂,代替实施例一中步骤(1)的柠檬酸,进行类似的磁性纳米粒子的表面修饰,即得具有不同功能团的表面修饰的磁性纳米粒子。
(2)表面固载大分子的磁性纳米粒子的制备:将需要表面固载的大分子,包括聚合物大分子、用化学键连接的嵌段聚合物、蛋白质、DNA、多肽、多糖等代替本实施例的(1)中的小分子功能化试剂,用类似的方法,即得表面固载大分子的磁性纳米粒子。
(3)大分子交联的磁珠的制备:将大分子包括聚合物大分子、用化学键连接的嵌段聚合物、蛋白质、DNA、多肽、多糖等代替实施例一中步骤(2)的聚合物PEI,按照相似的方法即可制备不同大分子交联的磁珠。
实施例三
本实施例提供的一种磁珠的制备方法与实施例一所述的磁珠的制备方法基本相同,区别之处在于:
(1)包埋PEI的磁性纳米粒子制备:取80mL的三蒸水于250mL的四颈瓶中,通氮气除氧气20min后,连接实验装置,检查气密性,保证气密性良好;将水浴温度调至35℃,机械搅拌的转速调至300rpb,保持四颈瓶中的氮气环境,准确称取4.05g的FeCl3·6H2O和1.98g的FeCl2·4H2O,加入到四颈瓶中搅拌溶解20min,在一定浓度的氨水溶液中加入一定量的聚合物大分子PEI(Mw=10000),将机械搅拌的转速调至600rpb,缓慢滴加氨水稀释液直至溶液pH=11,停止滴加氨水;将水浴温度升至60℃后继续反应1h。
反应结束后,将黑色的悬浮液转移至烧杯中,磁分离,弃上清液,用三蒸水复溶洗涤,磁分离,弃上层清液,如此重复,直至溶液呈中性;将得到的Fe3O4纳米粒分散在100mL的三蒸水中超声30min,静置即得包埋PEI的磁性纳米粒子。
(2)磁性纳米粒子的PEI交联制备磁珠:采用EDC·HCl和sulfo-NHS活化聚丙烯酸中的羧基,在37℃的条件下摇床活化1h,将其加入一定量的包埋PEI的磁性纳米粒子的水分散液中超声分散1min,37℃水浴条件下置于摇床中振摇反应1h,磁分离,水洗,复溶于水中,即得磁珠。
另外,可将含有大量活泼基团(包括羟基、羧酸、醛基、巯基和不同于PEI的其它聚合有机胺所含有的氨基)的大分子代替本实施例的(1)中的PEI,进行类似的大分子包埋,即得具有不同包埋大分子的磁性纳米粒子。
实施例四
本实施例提供了一种如实施例一或实施例二或实施例三所述的磁珠的制备方法制得的磁珠在生物医药分析与分离的应用,将磁珠直接加入生物样品中,利用电荷相互作用进行目标物的磁性快速捕获分离与分析;或者首先在磁珠上偶联捕获试剂,再进行目标物的磁性快速捕获分离与分析。
现以实施例三的表面带有羧基的磁珠为例说明。将50μL 5mg/mL的磁性用EDC-NHS方法活化羧基,磁分离,复溶于1.5mL蛋白低吸附的EP管中,加200ul0.01M pH7.4的PBS,涡旋使之均匀,加入50μL 40μg/mL兔抗小鼠IgG孵化30min,加入50μL的1%的BSA封闭30min,磁分离,加入300μL 0.01M pH7.4的PBS复溶,即得偶联捕获试剂兔抗小鼠IgG抗体的免疫磁珠。
对于被捕获组分生物反应特别是免疫反应来说,本发明制得的小体积磁珠所导致的空间位阻显著降低,捕获能力和分析检测灵敏度提高,能够更好地用于生物医药分离与分析,特别适用于生物体液或反应液中细胞、细菌、病毒、DNA/RNA、蛋白质等的分离以及基于化学发光、电化学发光和荧光的临床生化快速自动化检测。
本发明表面功能化的磁性纳米粒子的粒径范围为5-250nm,所述表面功能化的磁性纳米粒子可以为表面修饰的磁性纳米粒子或包埋大分子的磁性纳米粒子或表面固载大分子的磁性纳米粒子。磁珠的粒径至少为磁性纳米粒子粒径的2倍,具体范围为200nm-50μm,即动态光散射法测定的磁珠的水合粒径为200nm以上,最大可达50μm,且磁珠的形态为球形或不规则形。磁珠在溶液中的完全分离时间控制在1min-6min范围,即磁珠的快速分离时间最短为1min。
本发明利用交联剂的活泼基团和至少两个磁性纳米粒子的表面功能基团之间的化学反应所形成的化学键,将多个磁性纳米粒子连接起来,制备出高磁含量(70%-98%)的磁珠。
本发明制备方法简单,制备产率高,磁珠粒径大小可控;在制备磁珠中固载相同量的磁性纳米粒,即每个磁珠具有相同磁力的前提下,本发明制备的磁珠的磁含量更高,体积更小,表面电荷丰富,表面亲水性更强,悬浮稳定性更好,解决了现有磁珠特别是医用磁珠磁含量低的问题,也克服了现有磁珠为了实现快速分离,极大地增加磁珠体积而导致悬浮稳定性降低的问题。
以上结合具体实施例描述了本发明的技术原理。这些描述只是为了解释本发明的原理,而不能以任何方式解释为对本发明保护范围的限制。基于此处的解释,本领域的技术人员不需要付出创造性的劳动即可联想到本发明的其它具体实施方式,这些方式都将落入本发明的保护范围之内。
一种磁珠的制备方法及应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0