





专利摘要
本发明提供了一种酶催化的双迈克尔连续加成方法。上述加成方法包括:在连续化反应设备中,将一级胺类化合物和α,β不饱和酯在酶催化剂的催化作用下进行迈克尔双加成反应,形成三级胺产物;其中,一级胺类化合物为烷基、苯基取代的烷基、烷氧基或芳香基取代的胺。本发明提供的酶催化的双迈克尔连续加成方法有效解决了现有技术中迈克尔加成反应时间较长、需要贵金属催化剂、后处理复杂等问题。
权利要求
1.一种酶催化的双迈克尔连续加成方法,其特征在于,所述方法包括:在连续化反应设备中,将一级胺类化合物和α,β不饱和酯在酶催化剂的催化作用下进行迈克尔双加成反应,形成三级胺产物;其中,所述一级胺类化合物为烷基、苯基取代的烷基、烷氧基或芳香基取代的胺;所述酶催化剂选自脂肪酶CALB、PSSD、ITLIM、诺维信435、Mucor miehe中的一种或多种。
2.根据权利要求1所述的酶催化的双迈克尔连续加成方法,其特征在于,所述一级胺类化合物具有以下通式I、通式II或通式III所示结构:
通式I 通式II通式III
其中,n为1~4的整数,R选自C
3.根据权利要求1所述的酶催化的双迈克尔连续加成方法,其特征在于,所述一级胺类化合物选自苄胺、苯乙胺、对甲基苄胺、间甲基苄胺、邻甲基苄胺、对甲基苯乙胺、间甲基苯乙胺、邻甲基苯乙胺、甲氧基胺、乙氧基胺、甲胺、乙胺中的一种或多种。
4.根据权利要求1至3中任一项所述的酶催化的双迈克尔连续加成方法,其特征在于,所述α,β不饱和酯选自丙烯酸甲酯、甲基丙烯酸甲酯、丙烯酸乙酯、丙烯酸异丙酯、丙烯酸丁酯中的一种或多种。
5.根据权利要求1至3中任一项所述的酶催化的双迈克尔连续加成方法,其特征在于,所述方法包括以下步骤:
将所述一级胺类化合物溶解于第一溶剂中,形成第一原料液;
将所述α,β不饱和酯、所述酶催化剂溶解与第二溶剂中,形成第二原料液;
将所述第一原料液和所述第二原料液连续通入所述连续化反应设备中进行所述迈克尔双加成反应,得到所述三级胺产物。
6.根据权利要求5所述的酶催化的双迈克尔连续加成方法,其特征在于,所述第一溶剂和所述第二溶剂分别独立地选自醇类溶剂。
7.根据权利要求6所述的酶催化的双迈克尔连续加成方法,其特征在于,所述醇类溶剂为甲醇、乙醇、异丙醇中的一种或多种。
8.根据权利要求6所述的酶催化的双迈克尔连续加成方法,其特征在于,所述连续化反应设备为盘管反应器。
9.根据权利要求5所述的酶催化的双迈克尔连续加成方法,其特征在于,所述一级胺类化合物与所述α,β不饱和酯的摩尔比为1:2~1:4,所述酶催化剂的重量为所述一级胺类化合物重量的5~10%。
10.根据权利要求5所述的酶催化的双迈克尔连续加成方法,其特征在于,所述第一原料液中所述一级胺类化合物的浓度为0.1~0.5g/ml;所述第二原料液中所述α,β不饱和酯的浓度为0.4~0.8g /ml,所述酶催化剂的浓度为0.01~0.05g/ml。
11.根据权利要求5所述的酶催化的双迈克尔连续加成方法,其特征在于,在通入所述连续化反应设备的过程中,所述第一原料液和所述第二原料液的进料速度之比为1:1~1:2。
12.根据权利要求11所述的酶催化的双迈克尔连续加成方法,其特征在于,所述连续化反应设备中物料的保留时间为20~60min。
13.根据权利要求1至3中任一项所述的酶催化的双迈克尔连续加成方法,其特征在于,所述迈克尔双加成反应的温度为80~120℃。
说明书
技术领域
本发明涉及有机合成技术领域,具体而言,涉及一种酶催化的双迈克尔连续加成方法。
背景技术
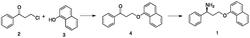
迈克尔加成反应是在碱催化下能提供亲核负碳离子的化合物和一个亲电共轭加成反应,是有机合成中增长碳链的常用方法之一。此反应为热力学控制的反应,通常加成反应的供体是活性亚甲基,反应的受体是活化烯烃。有文献(Journal of LabelledCompounds and Radiopharmaceuticals.2013,7,12,3110)报道以苄胺为原料与丙烯酸甲酯反应,以甲醇为溶剂,加热回流,反应过夜,制备双加成的叔胺,反应时间较长(18 h),甲醇回流放大反应存在安全隐患。
另有文献报道(Asian Journal of Chemistry; Vol. 23, No. 9 (2011), 3792-3794),以丙烯酸甲酯为原料与苯胺反应,PEG2000为溶剂,氯化铑为催化剂制备双迈克尔加成产物,反应时间长,温度高,后处理产生的废液量较大,但该方法使用贵金属催化剂,使放大生产后成本较高。
文献报道的方法,反应所需要的时间太长,催化剂后处理对环境造成的污染严重,后处理所涉及的成本较高,同时反应所需要的温度高,使用的溶剂甲醇形成蒸汽,放大生产存在较大的安全隐患,所以传统方法不具有竞争性。因此,需要开发一种绿色,高效简捷的合成的放大生产方式。
发明内容
本发明的主要目的在于提供一种酶催化的双迈克尔连续加成方法,以解决现有技术中迈克尔加成反应时间较长、需要贵金属催化剂、后处理复杂等问题。
为了实现上述目的,根据本发明的一个方面,提供了一种酶催化的双迈克尔连续加成方法,其包括:在连续化反应设备中,将一级胺类化合物和α,β不饱和酯在酶催化剂的催化作用下进行迈克尔双加成反应,形成三级胺产物;其中,一级胺类化合物为烷基、苯基取代的烷基、烷氧基或芳香基取代的胺。
进一步地,一级胺类化合物具有以下通式I、通式II或通式III所示结构:
通式I 通式II通式III
其中,n为1~4的整数,R选自C1至C6的烷基。
进一步地,一级胺类化合物选自苄胺、苯乙胺、对甲基苄胺、间甲基苄胺、邻甲基苄胺、对甲基苯乙胺、间甲基苯乙胺、邻甲基苯乙胺、甲氧基胺、乙氧基胺、甲胺、乙胺中的一种或多种。
进一步地,α,β不饱和酯选自丙烯酸甲酯、甲基丙烯酸甲酯、丙烯酸乙酯、丙烯酸异丙酯、丙烯酸丁酯中的一种或多种,优选α,β不饱和酯为丙烯酸甲酯。
进一步地,酶催化剂选自脂肪酶CALB、PSSD、ITLIM、诺维信435、Mucor miehe中的一种或多种,优选酶催化剂为脂肪酶CALB。
进一步地,方法包括以下步骤:将一级胺类化合物溶解于第一溶剂中,形成第一原料液;将α,β不饱和酯、酶催化剂溶解与第二溶剂中,形成第二原料液;将第一原料液和第二原料液连续通入连续化反应设备中进行迈克尔双加成反应,得到三级胺产物;优选地,第一溶剂和第二溶剂分别独立地选自醇类溶剂,优选醇类溶剂为甲醇、乙醇、异丙醇中的一种或多种,更优选醇类溶剂为甲醇;优选地,连续化反应设备为盘管反应器。
进一步地,一级胺类化合物与α,β不饱和酯的摩尔比为1:2~1:4,酶催化剂的重量为一级胺类化合物重量的5~10%。
进一步地,第一原料液中一级胺类化合物的浓度为0.1~0.5g/ml;第二原料液中α,β不饱和酯的浓度为0.4~0.8g /ml,酶催化剂的浓度为0.01~0.05g/ml。
进一步地,在通入连续化反应设备的过程中,第一原料液和第二原料液的进料速度之比为1:1~1:2;优选地,连续化反应设备中物料的保留时间为20~60min,更优选为30min。
进一步地,迈克尔双加成反应的温度为80~120℃,优选为100℃。
本发明提供的酶催化的双迈克尔连续加成方法,其是在连续化反应设备中将一级胺类化合物和α,β不饱和酯在酶催化剂的催化作用下进行迈克尔双加成反应。使用酶催化技术,避免了贵金属催化剂的使用,反应效率更高。尤其是将酶催化技术和连续化反应技术相结合,更是大大缩短了反应时间,且降低了反应温度,并极大地提高了反应的转化效率。酶作为催化剂,后处理操作简单,极大的降低了该产品的合成成本,同时降低后处理产生的废液,实现了成本控制和环境保护的双赢。连续化设备的使用,大大节约了人力成本,有益于工业放大生产。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
正如背景技术部分所描述的,现有技术中迈克尔加成反应时间较长、需要贵金属催化剂、后处理复杂等问题。
为了解决上述问题,本发明提供了一种酶催化的双迈克尔连续加成方法,其包括:在连续化反应设备中,将一级胺类化合物和α,β不饱和酯在酶催化剂的催化作用下进行迈克尔双加成反应,形成三级胺产物;其中,一级胺类化合物为烷基、苯基取代的烷基、烷氧基或芳香基取代的胺。
本发明提供的酶催化的双迈克尔连续加成方法,其是在连续化反应设备中将一级胺类化合物和α,β不饱和酯在酶催化剂的催化作用下进行迈克尔双加成反应。使用酶催化技术,避免了贵金属催化剂的使用,反应效率更高。尤其是将酶催化技术和连续化反应技术相结合,更是大大缩短了反应时间,且降低了反应温度,并极大地提高了反应的转化效率,收率能够达到大于95%的水平。酶作为催化剂,后处理操作简单,极大的降低了该产品的合成成本,同时降低后处理产生的废液,实现了成本控制和环境保护的双赢。连续化设备的使用,大大节约了人力成本,有益于工业放大生产。总之,该连续工艺结合酶催化技术,展现出绿色、高效、经济、安全的特性。
在一种优选的实施方式中,一级胺类化合物具有以下通式I、通式II或通式III所示结构:
通式I 通式II通式III
其中,n为1~4的整数,R选自C1至C6的烷基。
以上述结构的一级胺类化合物和α,β不饱和酯在酶催化剂的催化作用下进行迈克尔双加成反应,一方面具有更高的反应活性,反应效率更高;另一方面其与α,β不饱和酯、酶催化剂形成的反应体系早连续化反应设备中具有更好的稳定性。更优选地,一级胺类化合物选自苄胺、苯乙胺、对甲基苄胺、间甲基苄胺、邻甲基苄胺、对甲基苯乙胺、间甲基苯乙胺、邻甲基苯乙胺、甲氧基胺、乙氧基胺、甲胺、乙胺中的一种或多种。
在一种优选的实施方式中,α,β不饱和酯选自丙烯酸甲酯、甲基丙烯酸甲酯、丙烯酸乙酯、丙烯酸异丙酯、丙烯酸丁酯中的一种或多种,优选α,β不饱和酯为丙烯酸甲酯。选用以上几种α,β不饱和酯,具有更高的反应活性。
在一种优选的实施方式中,上述酶催化剂选自脂肪酶CALB、PSSD(Lipase PS“Amano” SD, Lipase from Burkholderia cepacia)、ITLIM(Lipozyme® TL IM, lipasefrom Thermomyces lanuginosus)、诺维信435、Mucor miehe中的一种或多种。这几种酶催化剂用于催化上述双迈克尔加成反应,具有更高的催化活性,有利于进一步提高反应效率,相应缩短反应时间,提高产品收率。更优选酶催化剂为脂肪酶CALB。
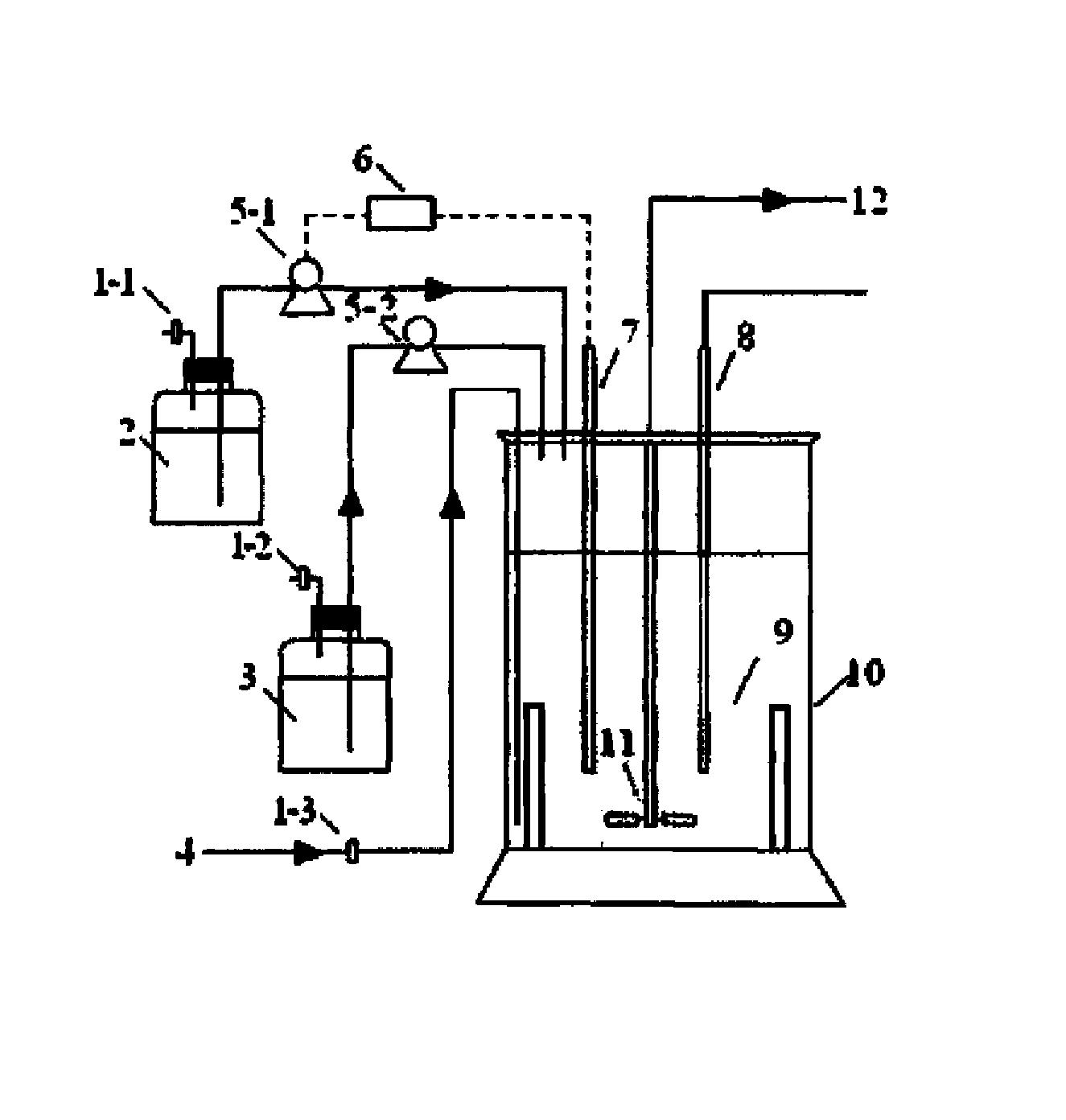
出于进一步;连续化反应稳定性的目的,在一种优选的实施方式中,上述方法包括以下步骤:将一级胺类化合物溶解于第一溶剂中,形成第一原料液;将α,β不饱和酯、酶催化剂溶解与第二溶剂中,形成第二原料液;将第一原料液和第二原料液连续通入连续化反应设备中进行迈克尔双加成反应,得到三级胺产物。这样,分别将一级胺类化合物、α,β不饱和酯和酶催化剂配制成不同体系的原料液,然后将两种原料液连续通入连续化反应设备,随着物料向前一边汇流一边反应,使得反应过程更为稳定、高效,且该过程中因物料连续进入产品连续排出,极大地减少了副反应的发生几率。
为了进一步提高反应稳定性,优选地,第一溶剂和第二溶剂分别独立地选自醇类溶剂,优选醇类溶剂为甲醇、乙醇、异丙醇中的一种或多种,更优选醇类溶剂为甲醇;更优选地,连续化反应设备为盘管反应器。更优选地,上述连续化反应设备还包括自动进料系统,电子秤、柱塞泵,隔膜泵,蠕动泵等,在实际操作过程中,可利用电子秤配料后,采用自动进料系统、柱塞泵,隔膜泵,蠕动泵等将反应原料液连续送入盘管反应器中进行连续化反应。
在一种优选的实施方式中,一级胺类化合物与α,β不饱和酯的摩尔比为1:2~1:4,酶催化剂的重量为一级胺类化合物重量的1~10%。更优选地,第一原料液中一级胺类化合物的浓度为0.1~0.5g/ml;第二原料液中α,β不饱和酯的浓度为0.4~0.8g/ml,酶催化剂的浓度为0.01~0.05g/ml。这样,反应过程中反应底物能够与酶催化剂更充分地接触并发生反应。
在一种优选的实施方式中,在通入连续化反应设备的过程中,第一原料液和第二原料液的进料速度之比为1:1~1:2。这样更有利于提高反应效率和产品收率。优选地,连续化反应设备中物料的保留时间为20~60min,更优选为30min。在该反应条件下,反应收率更高,
如前文所述,采用酶催化技术和连续化反应技术相结合,能够促使上述双迈克尔加成反应的条件更为温和,优选地,上述迈克尔双加成反应的温度为80~120℃,优选为100℃。干反应下,能够在保证较高的反应效率的基础上进一步降低产物纯度,减少副反应发生。
总之,采用本发明提供的上述工艺,取得了以下有益效果:
(1)首次实现了使用连续设备,以一级胺衍生物为原料,通过连续化反应,高效制备双迈克尔加成产物;
(2)连续反应模式,使反应时间缩短,极大提高了反应效率;
(3)酶作为连续反应的催化剂,极大提高了反应的转化效率。
(4)酶作为催化剂,后处理操作简单,极大的降低了该产品的合成成本,同时降低后处理产生的废液,实现了成本控制和环境保护的双赢;
(5)连续化设备的使用,大大节约了人力成本,有益于工业放大生产。
以下结合具体实施例对本申请作进一步详细描述,这些实施例不能理解为限制本申请所要求保护的范围。
实施例1
具体操作如下:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL甲醇中,原料液B为120g (3.0equiv)丙烯酸甲酯、50ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于50ml甲醇中;泵1以1.11g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.82g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物129.1g,收率97.5%。
参照上述工艺条件,底物合成实现1Kg级别放大,收率97%,重复性好。
实施例2
与上述实施例1不同之处在于,所选胺为对甲基苄胺:
原料液A 为56.5g(1.0equiv.)对甲基苄胺溶解于100mL甲醇中,原料液B为120g(3.0equiv)丙烯酸甲酯、50ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于50ml甲醇中;泵1以1.11g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.82g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物127.9g,收率92%。
实施例3
与实施例1不同之处在于,所用酯为丙烯酸丁酯:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL乙醇中,原料液B为178.6g (3.0equiv)丙烯酸甲酯、50ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于50ml甲醇中;泵1以1.11g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.82g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物156.8g,收率91%。
实施例4
与上述实施例1不同之处在于,所选酶为诺维信435:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL乙醇中,原料液B为120g (3.0equiv)丙烯酸甲酯、50ml(10%wt)诺维信435溶液(酶含量10%)溶解于50ml甲醇中;泵1以1.11g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.82g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物121.8,收率92%。
实施例5
与上述实施例1不同之处在于,所选甲醇用量为4V:
原料液A 为50g(1.0equiv.)苄胺溶解于150mL乙醇中,原料液B为120g (3.0equiv)丙烯酸甲酯,50ml(10%wt)诺维信435溶液(酶含量10%)溶解于50ml甲醇中;泵1以1.52g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.82g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物127.1,收率96%。
实施例6
与上述实施例1不同之处在于,所选酶催化剂浓度为0.01g/ml:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL甲醇中,原料液B为120g (3.0equiv)丙烯酸甲酯、5ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于50ml甲醇中;泵1以1.11g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.82g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物129.1g,收率97.5%。
实施例7
与上述实施例1不同之处在于,所选酯用量为4.0eq:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL甲醇中,原料液B为160g (4.0equiv)丙烯酸甲酯、50ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于50ml甲醇中;泵1以1.33g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.82g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物128.4g,收率97%。
实施例8
与上述实施例1不同之处在于,所述第二原料液中所述α,β不饱和酯的浓度为0.8g/ml:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL甲醇中,原料液B为160g (4.0equiv)丙烯酸甲酯、30ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于20ml甲醇中;泵1以1.11g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.95g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物124.5g,收率94.2%。
实施例9
与上述实施例1不同之处在于,所述两股料进料速度比为1:2:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL甲醇中,原料液B为120g (3.0equiv)丙烯酸甲酯、50ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于80ml甲醇中;泵1以1.11g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以2.22g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物122.01g,收率92.2%。
实施例10
与上述实施例1不同之处在于,保留时间为60 min:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL甲醇中,原料液B为120g (3.0equiv)丙烯酸甲酯、50ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于80ml甲醇中;泵1以0.55g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以0.975g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴100℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物120.4g,收率90.8%。
实施例11
与上述实施例1不同之处在于,反应温度为120 ℃:
原料液A 为50g(1.0equiv.)苄胺溶解于100mL甲醇中,原料液B为120g (3.0equiv)丙烯酸甲酯、50ml(10%wt)脂肪酶CALB酶溶液(酶含量10%)溶解于50ml甲醇中;泵1以1.11g/min的速度将原料液A泵入φ3 316L型不锈钢盘管中,泵2以1.82g/min的速度将原料液B泵入盘管φ3 316L型不锈钢盘管中;泵1、2同时开始打料,盘管置于外浴120℃外浴中,保留时间30min,盘管备压1.0~2.0 MPa,出料口直接连接放有1000 mL 四口瓶中,打料完毕后体系分液,取下层有机相,直接减压浓缩除去甲醇,得到无色油状物118.9g,收率90%。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
酶催化的双迈克尔连续加成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0