

IPC分类号 : C07D321/00,C07D321/12,C07D405/12,A61K31/625,A61P35/00,A61P31/04,A61P31/10,C12P17/08,C12P17/16,C12R1/465




专利摘要
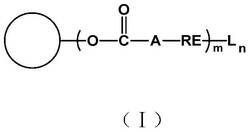
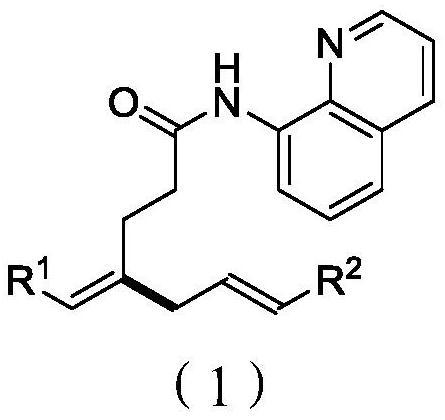
本发明涉及抗霉素类似物及其制备方法和用途,具体地公开了一类新型的抗霉素类似物,其结构如式Ⅰ或式Ⅱ所示,式中,R1、R2和R3同说明书中所定义。本发明还公开了该抗霉素类似物的制备方法和用途。本发明的抗霉素类似物的抗真菌活性提高了1倍,而细胞毒性提高了66倍。
权利要求
1.一种抗霉素类似物,其结构如式Ⅰ或式Ⅱ所示:
式中,
R1选自:取代或未取代的C1-10烷基、-C1-10亚烷基-C≡CH,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:卤原子、苯基、C6-8环烷基;
R2为-CO-R4、-CO-CH=CH-R5或-CO-CH=C-R6R7,式中,R4选自:苯基、吡啶基、取代或未取代的C1-10烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:卤原子、苯基、羟基、羧基、乙炔基;R5选自:苯基、取代或未取代的C1-10烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:卤原子、苯基、羟基、羧基、乙炔基;R6、R7各自独立地为C1-10烷基;
R3为H或卤原子;
附加条件是,所述抗霉素类似物不包括下列化合物:
2.如权利要求1所述的抗霉素类似物,其特征在于,所述R1选自:取代或未取代的C1-8烷基、-C1-6亚烷基-C≡CH,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:卤原子、苯基、C6-8环烷基。
3.如权利要求1所述的抗霉素类似物,其特征在于,所述R2为-CO-R4、-CO-CH=CH-R5或-CO-CH=C-R6R7;
式中,R4选自:苯基、吡啶基、C1-8烷基、取代的C1-5烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:氟原子、苯基、羟基、羧基、乙炔基;
R5选自:苯基、C1-8烷基、取代的C1-3烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:氟原子、苯基、羟基、羧基、乙炔基;
R6、R7各自独立地为C1-5烷基。
4.一种药物组合物,其特征在于,所述组合物包括:
如权利要求1所述的抗霉素类似物或其药学上可接受的盐;以及
药学上可接受的载体。
5.如权利要求1所述的抗霉素类似物的用途,其特征在于,用于制备:
(a)抗菌的组合物;
(b)抑制微生物生长的组合物;
(c)细胞生物学工具的药物;或
(d)抗癌药物。
6.一种体外非治疗性地抑制微生物生长或者杀灭微生物的方法,其特征在于,包括步骤:在需要处理的场所施用权利要求1所述的抗霉素类似物或权利要求4所述的药物组合物。
7.一种用于制备权利要求1所述抗霉素类似物的中间体,其特征在于,所述中间体如式Ⅲ或式Ⅳ所示:
式中,R1和R3同权利要求1中所定义,
附加条件是,所述中间体不包括下列化合物:
8.一种用于生产权利要求7所述中间体的链霉菌(Streptomyces sp.)突变菌株,其特征在于,所述菌株中的抗霉素生物合成基因簇中的酰基转移酶基因antB敲除或失活。
9.如权利要求7所述的中间体的制备方法,其特征在于,包括步骤:
(a)在合适表达条件下,在式Ⅶ所示的化合物和6-氟色氨酸的存在或不存在下,培养权利要求8所述的链霉菌(Streptomyces sp.)突变菌株,从而产生权利要求7所述的中间体,
R1-COOH 式Ⅶ
式中,R1同权利要求1中所定义;
(b)从发酵产物中分离出所述的中间体。
10.如权利要求1所述的抗霉素类似物的制备方法,其特征在于,包括步骤:
在酰基转移酶AntB的存在下,将式Ⅴ所示的化合物与权利要求8所述的中间体进行反应,从而得到权利要求1所述的抗霉素类似物,
CoA-S-R2 式Ⅴ
式中,R2同权利要求1中所定义。
说明书
技术领域
本发明属于生物技术工程领域,具体地涉及使用链霉菌重组菌株AL2110,通过前体喂养发酵制备脱酰基抗霉素类似物,以及在酰基转移酶AntB的存在下制备抗霉素类似物的方法。
背景技术
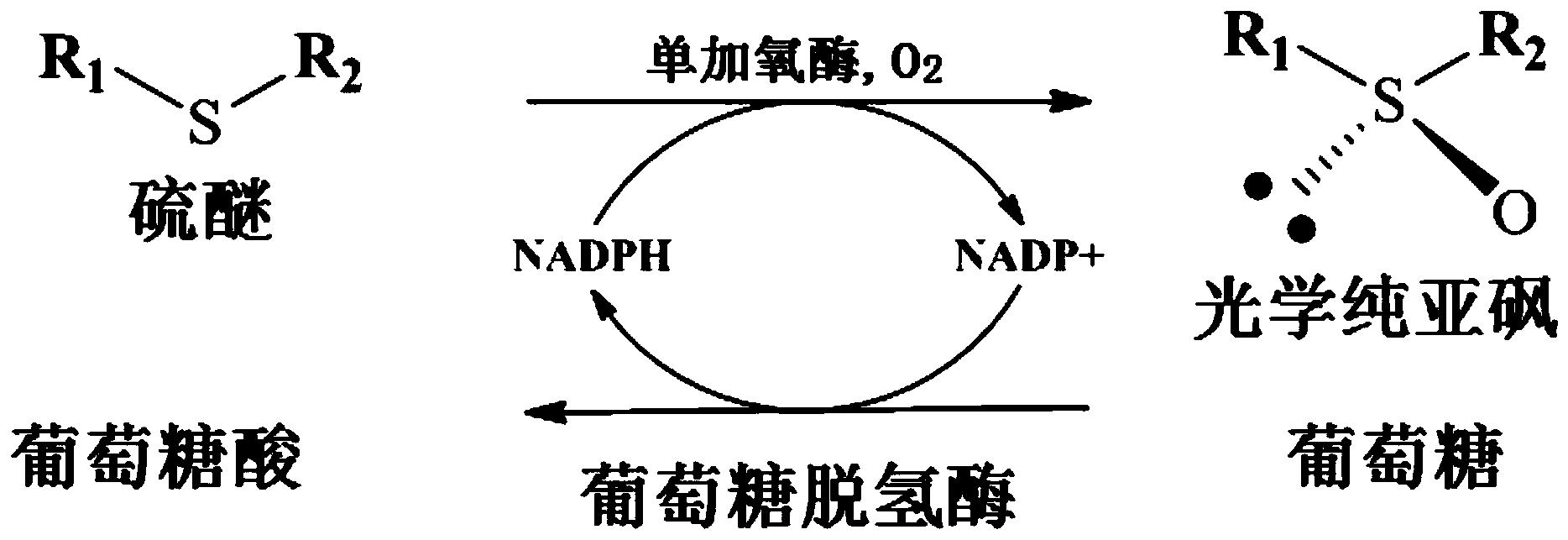
抗霉素是九元环二内酯类抗生素,其九元环上通过酰胺键连接3-甲酰胺基水杨酸,和两个具有多种变化的取代基团R1和R2。目前所报道的抗霉素类似物根据R1和R2基团的不同组合已有44种。它们都是由链霉菌产生,并具有广谱的抗真菌活性和诱导细胞凋亡的活性。目前抗霉素主要有两个用途,首先在生命科学研究中被作为工具药物用来抑制线粒体呼吸链电子传递,另外在渔业生产中被用来特异性的清除有害鱼类。
但是由于抗霉素天然组分复杂,化学性质相似,从产生菌中分离得到单一组分困难,因而目前人们通常将多种抗霉素组分的混合物进行使用,因此缺乏对于抗霉素单一组分生物学活性的认识。另外由于抗霉素结构的复杂性,采用化学合成的方法来生产其类似物操作繁琐,而且产率低下。因而对于抗霉素单一组分生物学活性的发掘和筛选受到了严重的阻碍。
发明内容
本发明的目的在于提供一类更高活性的抗霉素类似物,以及提供所述抗霉素类似物的制备方法和用途。
本发明第一方面提供了一种抗霉素类似物,其结构如式Ⅰ或式Ⅱ所示:
式中,
R1选自:取代或未取代的C1-10烷基、-C1-10亚烷基-C≡CH,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:卤原子、苯基、C6-8环烷基;
R2为-CO-R4、-CO-CH=CH-R5或-CO-CH=C-R6R7,式中,R4选自:苯基、吡啶基、取代或未取代的C1-10烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:卤原子、苯基、羟基、羧基、乙炔基;R5选自:苯基、取代或未取代的C1-10烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:卤原子、苯基、羟基、羧基、乙炔基;R6、R7各自独立地为C1-10烷基;
R3为H或卤原子;
附加条件是,所述抗霉素类似物不包括下列化合物:
在另一优选例中,所述R3为H或氟原子。
在另一优选例中,所述R1选自:取代或未取代的C1-8烷基、-C1-6亚烷基-C≡CH,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:卤原子、苯基、C6-8环烷基。
在另一优选例中,所述R1选自:C4-8烷基、-C3-6亚烷基-C≡CH、取代的C1-8烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:氯原子、苯基、环己基。
在另一优选例中,所述R1选自:C4-8烷基、-C3-6亚烷基-C≡CH、氯代的C2-4烷基、苯基取代的C1-3烷基、环己基取代的C1-3烷基。
在另一优选例中,所述R1为选自下组的基团:
在另一优选例中,所述R1选自下组基团:
在另一优选例中,所述R2为-CO-R4、-CO-CH=CH-R5或-CO-CH=C-R6R7;
式中,R4选自:苯基、吡啶基、C1-8烷基、取代的C1-5烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:氟原子、苯基、羟基、羧基、乙炔基;
R5选自:苯基、C1-8烷基、取代的C1-3烷基,其中,所述的“取代”指基团中一个或多个H被选自下组的取代基所取代:氟原子、苯基、羟基、羧基、乙炔基;
R6、R7各自独立地为C1-5烷基。
在另一优选例中,所述R4选自:苯基、吡啶基、C1-8烷基、取代的C1-5烷基,其中,所述的“取代”指基团中一个H被选自下组的取代基所取代:三氟甲基、苯基、羟基、羧基、乙炔基。
在另一优选例中,所述R5选自:苯基、C1-8烷基、氟代的C1-3烷基、乙炔基取代的C1-3烷基。
在另一优选例中,所述R5选自:苯基、C1-8烷基、三氟甲基取代的C1-3烷基、乙炔基取代的C1-3烷基。
在另一优选例中,所述R6、R7各自独立地为C1-3烷基。
在另一优选例中,所述R2为选自下组的基团:
在另一优选例中,所述R2为选自下组的基团:
在另一优选例中,所述抗霉素类似物选自下组:
本发明第二方面提供了一种药物组合物,所述组合物包括:
第一方面所述的抗霉素类似物或其药学上可接受的盐;以及
药学上可接受的载体。
本发明第三方面提供了第一方面所述抗霉素类似物的用途,用于制备:
(a)抗菌的组合物;
(b)抑制微生物生长的组合物;
(c)细胞生物学工具的药物;或
(d)抗癌药物。
在另一优选例中,所述的组合物为药物组合物。
在另一优选例中,所述细胞生物学工具药物是线粒体呼吸链抑制剂。
在另一优选例中,所述的微生物包括:细菌、真菌。
本发明第四方面提供一种体外非治疗性地抑制微生物生长或者杀灭微生物的方法,包括步骤:在需要处理的场所施用第一方面所述的抗霉素类似物或第二方面所述的药物组合物。
在另一优选例中,提供一种治疗动物细菌感染的方法,其特征在于,包括步骤:给需要治疗的哺乳动物施用第一方面所述的抗霉素类似物或第二方面所述的药物组合物。
本发明第五方面提供了一种用于制备第一方面所述抗霉素类似物的中间体,所述中间体如式Ⅲ或式Ⅳ所示:
式中,R1和R3同上所定义,
附加条件是,所述中间体不包括下列化合物:
本发明第六方面提供了一种用于生产第五方面所述中间体的链霉菌(Streptomyces sp.)突变菌株,所述菌株中的抗霉素生物合成基因簇中的酰基转移酶基因antB敲除或失活。
本发明第七方面提供了第五方面所述中间体的制备方法,包括步骤:
(a)在合适表达条件下,在式Ⅶ所示的化合物和6-氟色氨酸的存在或不存在下,培养第六方面所述的链霉菌(Streptomyces sp.)突变菌株,从而产生第五方面所述的中间体,
R1-COOH 式Ⅶ
式中,R1同权利要求1中所定义;
(b)从发酵产物中分离出所述的中间体。
在另一优选例中,所述R1为选自下组的基团:
本发明第八方面提供一种第一方面所述的抗霉素类似物的制备方法,包括步骤:
在酰基转移酶AntB的存在下,将式Ⅴ所示的化合物与第五方面所述的中间体进行反应,从而得到第一方面所述的抗霉素类似物,
CoA-S-R2 式Ⅴ
式中,R2同上所定义。
在另一优选例中,所述链霉菌突变菌株的制备方法包括:
(a)提供一含有抗霉素生物合成基因簇并能够产生抗霉素的菌株;
(b)将菌株中的酰基转移酶基因antB敲除或失活,得到所述的链霉菌突变菌株。
在另一优选例中,所述式Ⅴ所示的化合物的制备方法包括步骤:
将式Ⅵ所示的化合物与辅酶CoA-SH进行反应,从而得到式Ⅴ所示的化合物。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
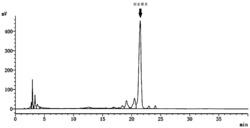
图1为脱酰基抗霉素的HPLC检测分析图。
具体实施方式
发明人经过广泛深入的研究,通过对抗霉素生物合成基因簇的克隆以及生物合成机制研究,利用突变生物合成、前体导向喂养和酶学转化方法,首次成功制备获得了一类新型的、活性更高的抗霉素类似物。在此基础上完成了本发明。
具体地,本发明提供了一种可产生脱酰基抗霉素的链霉菌突变菌株,该突变菌株中抗霉素生物合成基因簇中的酰基转移酶基因antB敲除。通过在发酵培养过程中添加(或喂养)抗霉素生物合成前体,从而获得对应的脱酰基抗霉素类似物。将制得的脱酰基抗霉素类似物在酰基转移酶AntB的存在下,与具有不同取代基的辅酶A进行反应,从而获得相对应的抗霉素类似物。
实验证明,本发明的抗霉素类似物抗真菌活性和细胞毒性方面有所提高,抗真菌活性提高了1倍,而细胞毒性提高了66倍。
基团定义
术语“C1-10烷基”指具有1-10个碳原子的直链或支链烷基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、己基、庚基、辛基、壬基、癸基或类似基团。
术语“C6-8环烷基”指具有6-8个碳原子的环状烷基,包括环己基、环庚基、环辛基。
术语“卤素原子”和“卤原子”均指氟原子、氯原子、溴原子、或碘原子。
术语“卤代的”指被相同或不同的一个或多个上述卤素原子取代的基团,例如三氟甲基、五氟乙基、或类似基团。
如本文所用,术语“链霉菌突变菌株”、“突变菌株”和“重组菌株”可互换使用,均指可生产脱酰基抗霉素类似物的链霉菌(Streptomyces sp.)突变菌株,其中,菌株中抗霉素生物合成基因簇中的酰基转移酶基因(antB)敲除。
如本文所用,术语“式I所示化合物”、“式I化合物”或“式I所示的化合物”可互换使用,均代表式I所示结构的化合物。
如本文所用,术语“本发明的抗霉素类似物”、“抗霉素类似物”均指本发明式I或式II所示的化合物。
如本文所用,术语“本发明的中间体”、“中间体”和“脱酰基抗霉素类似物”可互换使用,均指本发明式Ⅲ或式Ⅳ所示的化合物。
如本文所用,术语“抗霉素合成砌块”和“辅酶A衍生物”均指本发明式Ⅴ所示的化合物。
本发明所用的原料或试剂除特别说明之外,均市售可得。
酰基转移酶AntB
本发明所述酰基转移酶AntB是指编码序列如SEQ ID NO.:1所示,或氨基酸序列如SEQ ID NO.:2所示的酶蛋白。其主要用于催化酰基转移,形成酯或酰胺的酶,酰基的供体大多是酰基辅酶A。
出发菌株
本发明可以用任何含有抗霉素生物合成基因簇、能够产生抗霉素的菌株为出发菌株。例如,目前常用的产生抗霉素的菌株包括(但并不限于):Streptomyces blastmyceticus NBRC12747,Streptomyces albus J1074,Streptomyces ambofaciens ATCC 23877和Streptomyces sp.S4。
在本发明的一个优选例中,出发菌株是编号为Streptomyces sp.NRRL2288的链霉菌(Streptomyces sp.)。购于德国微生物菌种保藏中心(DSMZ),保存在美国农业研究菌种保藏中心(NRRL)。应理解,出发菌株不仅包括编号为Streptomyces sp.NRRL2288的菌株,还包括其衍生菌株。
链霉菌突变菌株及其构建方法
本发明的链霉菌突变菌株是一种可生产脱酰基抗霉素类似物的链霉菌(Streptomyces sp.)突变菌株,其中,菌株中抗霉素生物合成基因簇中的酰基转移酶基因(antB)敲除。所述的突变菌株可以通过对出发菌株采用基因敲除或失活的方法(例如通过同框缺失的手段)而构建。
在本发明的一个优选例中,提供了一种构建可产生脱酰基抗霉素突变株的方法,它包括基因同框缺失质粒的构建,将质粒导入野生型菌株,通过同源臂与野生型菌株基因组的重组,从而从基因组中敲除相应基因,并获得基因同框缺失或失活的突变株。缺失抗霉素生物合成基因簇中的酰基转移酶基因(antB)的突变株无法正常合成抗霉素,只生产脱酰基抗霉素。
具体地,包括步骤:
1.构建基因antB的同框缺失质粒
PCR扩增用于基因敲除的antB基因的左臂和右臂片段,将这两个片段连入pKC1139中(具体可参考文献:Org.Lett.2012,14,4142–4145.),获得重组质粒。将重组质粒转化到E.coli DH5α(具体可参考文献:Org.Lett.2012,14,4142–4145.),挑取单克隆菌落扩增培养,并将扩增后验证正确的质粒转化到E.coli S17-1(具体可参考文献:Org.Lett.2012,14,4142–4145.)。
2.制备重组菌株
收集野生型的Streptomyces sp.NRRL2288的新鲜孢子,并用SET缓冲溶液(β-D-呋喃果糖基-α-D-吡喃葡萄糖苷(Sucrose)0.3M,Tris-HCl25mM,EDTA25mM,pH=8.0)清洗两次。清洗后的孢子重悬在500uLSET缓冲液中,放于50℃水浴中热激。热激后的孢子在30℃萌发3小时左右,与含有重组质粒的E.coliS17-1在MS平板(黄豆饼粉20g/L,甘露糖20g/L,琼脂糖20g/L,自来水配制)上按照一定比例混合涂抹,并30℃中培养12小时后用阿伯拉霉素覆盖平板。接合子在温箱培养2到3天后长出,挑选接合子到阿伯拉抗性平板上37℃培养2到3天。选取长势良好的接合子于TSB(无抗性)溶液(Tryptone Soya Broth 30g/L)中传代三次以上,再于平板上划线筛选单菌落。通过阿伯拉抗性筛选,选取不具有阿伯拉抗性的菌株进行发酵和PCR基因型验证以获得antB同框缺失的重组菌株。
中间体及其制备方法和用途
如本文所用,术语“中间体”指参与本发明的抗霉素类似物合成的化合物。这些中间体化合物被用于合成或制备本发明的抗霉素类似物。一类优选的中间体包括式DA-3、DA-5、DA-6、DA-9、DA-10、DA-11、DA-12、DA-13、DA-14、DA-15和DA-16所示的化合物。
在本发明的一个优选例中,提供了一种本发明第五方面所述的中间体的制备方法,通过发酵培养过程中喂养不同的抗霉素生物合成前体,从而获得对应的脱酰基抗霉素类似物。所述抗霉素生物合成前体如式Ⅶ所示。
具体地,包括以下步骤:
(1)DA-1,DA-2,DA-3,DA-4,DA-5和DA-6的制备
将antB缺失的突变菌株接种于1L发酵培养基中,所述培养基包括:可溶性淀粉10g,蛋白胨-B4g,酵母提取物2g和碳酸钙1g,用自来水定容至1L,pH=7.0,于28℃,250rpm发酵4天后处理发酵液和菌丝。
(2)DA-8,DA-9,DA-10,DA-11和DA-12的制备
将antB缺失的突变菌株接种于1L发酵培养基中,所述培养基包括:可溶性淀粉10g,蛋白胨-B4g,酵母提取物2g和碳酸钙1g,用自来水定容至1L,pH=7.0,于28℃,250rpm发酵1天后,于发酵液中分别加入肉桂酸(用于生产DA-8),5-氯戊酸(用于生产DA-9),环己基丙酸(用于生产DA-10)和10-十一烷炔酸(用于生产DA-11和DA-12)至终浓度1mM,而后继续发酵3天。
(3)DA-13,DA-14和DA-15的制备
将antB缺失的突变菌株接种于1L发酵培养基中,所述培养基包括:可溶性淀粉10g,蛋白胨-B4g,酵母提取物2g和碳酸钙1g,用自来水定容至1L,pH=7.0,于28℃,250rpm发酵1天后,于发酵液中加入6-氟色氨酸至终浓度1mM,而后继续发酵3天。
(4)DA-16的制备
将antB缺失的突变菌株接种于1L发酵培养基中,所述培养基包括:可溶性淀粉10g,蛋白胨-B4g,酵母提取物2g和碳酸钙1g,用自来水定容至1L,pH=7.0,于28℃,250rpm发酵1天后,于发酵液中分别加入环己基丙酸和6-氟色氨酸分别至终浓度1mM,而后继续发酵3天。
发酵结束后,将发酵液pH调至2.5~3.0,离心后上清弃去,菌体用甲醇:二氯甲烷=1:2的混合溶剂浸泡过夜,过滤除去不溶物,滤液经减压蒸馏抽干得深褐色膏状物。而后用甲醇溶解膏状物,经离心后弃去不溶物,将上清于HPLC进行半制备分离。
HPLC的半制备分离条件为:
仪器:Agilent 1100 HPLC系统
柱子:Agilent Zorbax SB-C18column(5μm,250×9.4mm)
检测波长:UV=242nm
流动相:A=水;B=乙腈
流速:3ml/min。
流动相梯度配比:T=0min,40%B;T=10min,40%B;T=35min,100%B;T=45min,100%B(A,水+0.1%甲酸;B,乙腈+0.1%甲酸)。
酰基转移酶AntB的制备
在本发明的一个优选例中,所述的酰基转移酶AntB的制备方法包括步骤:
1.构建酰基转移酶AntB的异源表达质粒
PCR扩增用于蛋白质表达的antB基因片段,将这个片段连入pSJ5(参考文献:现代食品科技2011,27,905–907,将pET32a的卡那霉素抗性改造为氨苄青霉素抗性获得,其中pET32a购自Merck KGaA,Darmstadt,Germany)中,获得重组质粒。将重组质粒转化到E.coli DH5α,挑取单克隆菌落扩增培养,并将扩增后并验证正确的质粒转化E.coli BL21(DE3)中,得到antB表达菌株AL2120。
2.酰基转移酶AntB的异源表达和纯化
将携带antB的异源表达质粒的菌株AL2120接种于LB(胰蛋白胨10g/L,酵母提取物5g/L,NaCl10g/L)中,于37℃,220rpm摇床培养至OD600=0.6,加入IPTG至终浓度为100mM。于16℃表达20小时后,5000rpm4℃离心,收集菌体。将菌体在破菌缓冲液(20mM Tris-HCl pH7.5,100mM NaCl,10%glycerol,5mM imidazole)中重悬,4℃超声破碎后,8000rpm离心,将上清液用镍柱分离并用TEV蛋白酶酶切得到AntB。
抗霉素合成砌块及制备方法
如本文所用,术语“抗霉素合成砌块”指参与本发明抗霉素类似物合成的化合物,其中所述的化合物为式Ⅴ所示的辅酶A衍生物。此化合物被用于合成或制备本发明的抗霉素类似物,一类优选的辅酶A衍生物选自下组:
在本发明的一个优选例中,所述的辅酶A衍生物的制备方法包括步骤:
将式Ⅵ所示的化合物与辅酶CoA-SH进行反应,从而得到所述的辅酶A衍生物
式中,R2同权利要求1中所定义。
式Ⅵ所示的化合物可采用常规的合成方法进行制备或从市场上购买。
抗霉素类似物及其制备方法和活性测试
本发明的抗霉素类似物为式Ⅰ或式Ⅱ所示的化合物,一类优选的化合物如下所示:
在本发明的一个优选实施例中,提供了一种本发明的抗霉素类似物的制备方法,包括步骤:
在酰基转移酶AntB的存在下,将辅酶A衍生物与脱酰基抗霉素类似物进行反应,从而得到本发明的抗霉素类似物。
反应路线如下式所示:
其中辅酶A衍生物的结构为CoA-S-R2,式中,R2同上述所定义。
通过测定细胞毒性(小鼠leukemia P388细胞系)和抗真菌活性(Candida albicans),最终发现本发明的抗霉素类似物均具有生物学活性,而且与标准品A3b(结构如下所示,购买于Sigma-Aldrich Co.LLC.)相比,其中约60%的类似物细胞毒性有所增强(66.7倍),约20%的类似物抗真菌活性有所增加(2倍)。
药物组合物和施用方法
本发明的抗霉素类似物具有优异的抑菌(抗菌)活性,因此可用作抗菌素(或抗生素)。
本发明还提供了一种组合物,它含有有效量的本发明的抗霉素类似物,以及药学上可接受的载体。
如本文所用,术语“有效量”或“有效剂量”是指可对人和/或动物产生功能或活性的且可被人和/或动物所接受的量,如0.001-99wt%;较佳的0.01-95wt%;更佳的,0.1-90wt%。
如本文所用,“药学上可接受的”的成分是适用于人和/或哺乳动物而无过度不良副反应(如毒性、刺激和变态反应)的,即具有合理的效益/风险比的物质。术语“药学上可接受的载体”指用于治疗剂给药的载体,包括各种赋形剂和稀释剂。
本发明的抗霉素类似物可施用于哺乳动物(如人),可以口服、直肠、肠胃外(静脉内、肌肉内或皮下)、局部等方式给药。所述抗霉素类似物可以单独给药,或者与其他药学上可接受的化合物联合给药。需要指出,本发明的抗霉素类似物可以混合给药。
本发明的药物组合物可根据治疗的需要,制成用于抗菌药物、抗癌药物的各类合适剂型。所述的药物组合物宜在无菌条件下制造。抗霉素类似物的给药量是治疗有效量。本发明的药物制剂还可制成缓释制剂。
使用药物组合物时,是将安全有效量的本发明的抗霉素类似物适用于需要治疗的哺乳动物(如人),其中施用时剂量为药学上认为的有效给药剂量,对于60kg体重的人而言,日给药剂量通常为1~1000mg,优选20~500mg。当然,具体剂量还应考虑给药途径、病人健康状况等因素,这些都是熟练医师技能范围之内的。
本发明的主要优点包括:
(1)提供了一类结构新颖且活性更高的抗霉素类似物。其中大部分抗霉素类似物的抗真菌活性提高了1倍,而细胞毒性提高了66倍。
(2)提供了一种制备中间体脱酰基抗霉素类似物以及抗霉素类似物方法。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如Sambrook等人,分子克隆:实验室手册(New York:Cold Spring Harbor Laboratory Press,1989)中所述的条件,或按照制造厂商所建议的条件。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明中。文中所述的较佳实施方法与材料仅作示范之用,但不能限制本发明的内容。
实施例1
链霉菌重组菌株AL2110的构建
1.构建antB同框缺失质粒
克隆antB同框缺失左臂的引物序列如下:
antB左臂正向引物:5’-AAAAAGCTTTGGAAGAGTTCTGGCGGACC-3’(SEQ ID NO.:3)
antB左臂反向引物:5’-AAATCTAGACTCCTTCGACAACCGGCTC-3’(SEQ ID NO.:4)
克隆tsrT同框缺失右臂的引物序列如下:
antB右臂正向引物:5’-AAATCTAGAAGTACCGAGGGGTCGACGTT-3’(SEQ ID NO.:5)
antB右臂反向引物:5’-AAAGAATTCAGAGCTGGGGGAGGGTACGT-3’(SEQ ID NO.:6)
以pAL2602(以pCC1-FOS为基础的粘粒,包含整个抗霉素生物合成基因簇,具体可参考文献Org.Lett.2012,14,4142–4145.)为模板,以dNTP,DMSO,无酶水,高保真的Primestar DNA聚合酶及缓冲液组成PCR反应体系,扩增用于antB基因敲除的左臂和右臂片段。
将克隆后的两个片段凝胶电泳分离,切胶回收并纯化,分别加入限制性内切酶HindIII和XbaI以及XbaI和EcoRI消化回收片段,将其连入用限制性内切酶HindIII和EcoRI同样酶处理过的pKC1139中,将连接体系转化到E.coliDH5α,挑取单克隆菌落于LB培养液(含有阿伯拉霉素抗生素)中培养过夜,至菌液较浓。提取质粒经酶切验证后送测序进一步验证。将验证正确的质粒转化E.coli S17-1中。
2.重组菌株AL2110的获得
收集野生型的Streptomyces sp.NRRL2288的新鲜孢子,并用SET缓冲溶液清洗两次。清洗后的孢子重悬在500uLSET缓冲液中,放于50℃水浴中热激。热激后的孢子在30℃萌发3小时左右,与含有重组质粒的E.coli S17-1在MS平板上按照一定比例混合涂抹,并30℃中培养12小时后用阿伯拉霉素覆盖平板。接合子在温箱培养2到3天后长出,挑选接合子到阿伯拉抗性平板上37℃培养2到3天。选取长势良好的接合子于TSB(无抗性)溶液中传代三次以上,再于平板上划线筛选单菌落。通过阿伯拉抗性筛选,选取不具有阿伯拉抗性的菌株进行发酵和PCR基因型验证以获得antB同框缺失的重组菌株AL2110。
实施例2 5-氟-7-环己基甲基脱酰基抗霉素DA-16的制备
将antB缺失的突变株AL2110接种于1L发酵培养基A1中(可溶性淀粉10g,蛋白胨-B4g,酵母提取物2g,碳酸钙1g,自来水定容至1L,pH=7.0)于28℃,250rpm发酵。在发酵1天后,于发酵液中分别加入环己基丙酸和6-氟色氨酸分别至终浓度1mM,而后继续发酵3天。
发酵结束后,将发酵液pH调至2.5~3.0,离心后上清弃去,菌体用甲醇:二氯甲烷=1:2的混合溶剂浸泡过夜,过滤除去不溶物,滤液经减压蒸馏抽干得深褐色膏状物。而后用甲醇溶解膏状物,经离心后弃去不溶物,将上清于HPLC进行半制备分离。
HPLC的半制备分离条件为:
仪器:Agilent1100HPLC系统
柱子:Agilent Zorbax SB-C18column(5μm,250×9.4mm)
检测波长:UV=242nm
流动相:A=水;B=乙腈
流速:3ml/min。
流动相梯度配比:T=0min,40%B;T=10min,40%B;T=35min,100%B;T=45min,100%B(A,水+0.1%甲酸;B,乙腈+0.1%甲酸).
按上述的HPLC的洗脱条件,收集流出液,最终得到目标产物。对目标产物进行鉴定,结果如下:
1H NMR(300MHz,CDCl3):δ12.41(s,1H),8.51(s,1H),8.42(dd,J=10.0Hz,J=2.6Hz,1H),8.03(s,1H),7.01(d,J=7.2,1H),6.95(dd,J=8.3Hz,J=2.7Hz,1H),5.70(m,1H),5.24(t,J=7.5Hz,1H),4.89(m,1H),3.57(t,J=9.3Hz,1H),2.48(m,1H),2.05(brs,1H),1.78~1.55(m,7H),1.46(d,J=6.2Hz,3H),1.31(d,J=6.7,3H),1.27~1.10(m,4H),0.96(m,1H),0.84(m,1H).13C NMR(75MHz,CDCl3):δ174.5,170.3,167.0,159.3,155.0(d,J=237.9Hz),147.2(d,J=1.5Hz),128.6(d,J=11.3Hz),113.3(d,J=29.4Hz),112.3(d,J=7.5Hz),105.7(d,J=24.4Hz),77.6,77.2,71.0,54.2,49.9,36.8,36.1,34.5,32.6,26.7,26.5,26.4,18.8,15.4.19F NMR(282MHz,CDCl3):δ-120.76(t,J=9.0Hz).HRMS(m/z):[M+H]+calcd.for C24H32FN2O8,495.2137;found,495.2132.
鉴定结果表明,成功制备了5-氟-7-环己基甲基脱酰基抗霉素DA-16。
实施例3 出发菌株和突变菌株的发酵产物分析
分别将野生型Streptomyces sp.NRRL2288和突变株AL2110进行发酵,发酵产物分别用HPLC进行分析,结果如图1所示:Ⅰ表示野生型Streptomyces sp.NRRL2288发酵,产生天然抗霉素组分A1(包括A1a和A1b)、A2(包括A2a和A2b)、A3(包括A3a和A3b)和A4(包括A4a和A4b);Ⅱ表示突变株AL2110发酵,产生脱酰基抗霉素DA-1,DA-2,DA-3,DA-4,DA-5和DA-6;Ⅲ表示突变株AL2110喂养肉桂酸发酵,产生脱酰基抗霉素DA-8;Ⅳ表示突变株AL2110喂5-氯戊酸发酵,产生脱酰基抗霉素DA-9;Ⅴ表示突变株AL2110喂养环己基丙酸发酵,产生脱酰基抗霉素DA-10;Ⅵ表示突变株AL2110喂养10-十一炔酸发酵,产生脱酰基抗霉素DA-11和DA-12;Ⅶ表示突变株AL2110喂养6-氟色氨酸发酵,产生脱酰基抗霉素DA-13,DA-14和DA-15;Ⅷ表示突变株AL2110同时喂养6-氟色氨酸和环己基丙酸发酵,产生脱酰基抗霉素DA-16。
结果表明,野生型菌株能够产生天然抗霉素组分,antB基因缺失突变株AL2110可以生产脱酰基抗霉素;而在antB基因缺失突变株AL2110喂养抗霉素生物合成砌块则可以产生R1,R2或R3单独或同时被修饰的脱酰基抗霉素。
实施例4 (2E)-2-庚烯-6-炔酰基辅酶A的制备
(2E)-2-庚烯-6-炔酰基辅酶A(化合物4)的制备方法,包括步骤:
(a)化合物1的制备
在冰浴下于50mL二氯甲烷中加入5.0g溴乙酰溴,一边搅拌一边逐滴加入10mL三乙胺。于室温下搅拌0.5h后,用1M盐酸淬灭反应。反应液经二氯甲烷萃取后,合并有机相,分别用NaHCO3和NaCl饱和水溶液洗三次,经柱层析分离得到11g S-溴乙酰基苯硫酚,即化合物1。核磁分析:1H NMR(400MHz,CDCl3):δ7.40~7.33(m,5H),4.05(s,1H).13C NMR(100MHz,CDCl3):δ191.0,134.5,130.0,129.5,126.8,33.2.
(b)化合物2的制备
将18g PPh3溶于20mL苯中,室温下逐滴加入9.0g化合物1。反应过程中体系出现大量白色沉淀。回流4h后,将反应液浓缩除去苯后溶解于热水中。将水相分离合并,而后与10%NaOH水溶液混合,并用氯仿萃取。分离合并有机相,经干燥浓缩后得到14g黄色粉末,即化合物2粗品。
(c)化合物3的制备
将1.0g4-戊炔醛和1.5当量化合物2溶于20mL甲苯中,室温搅拌48h后,浓缩并进行柱层析分离得到1.5g化合物3。核磁分析:1H NMR(400MHz,CDCl3):δ7.40~7.20(m,5H),6.72(dt,J=15.4Hz,7.2Hz,1H),6.26(d,J=15.4Hz,1H),2.24~2.19(m,2H),2.18~2.13(m,2H),1.89(t,J=3.5Hz,1H).13C NMR(100MHz,CDCl3):δ187.5,143.9,134.7,129.6,129.3,128.9,127.7,82.6,70.1,29.9,17.4.
(d)化合物4的制备
将5mg辅酶AcoA-SH溶于1mL磷酸缓冲液pH=8.5,而后加入1mL四氢呋喃和1.2当量化合物3,与室温搅拌过夜。反应完毕后,经浓缩除去四氢呋喃后,将水相用等体积乙醚洗3次,进行HPLC半制备分离,纯化得到1mg化合物4,即(2E)-2-庚烯-6-炔酰基辅酶A。高分辨质谱检测:HRMS(m/z):[M+H]+calcd.for C28H43N7O17P3S,874.1643;found,874.1650。
实施例5 制备抗霉素A1-19
在10mL pH=6.050mM MES(2-吗啉乙磺酸)缓冲液中加入5mM DA-1(8mg),10mM化合物4和1mg antB。于30℃反应2小时后,加入等体积乙酸乙酯淬灭反应,并将产物A1-19提取出来,即得到抗霉素A1-19。
实施例6 测定抗霉素类似物的抗真菌活性和细胞毒性
对抗霉素类似物进行抗菌活性的测定,方法如下:首先将测试菌C.albicans在含有4%葡萄糖和1%据蛋白胨的培养基中培养12小时后,将溶于二甲基亚砜的抗霉素类似物分别加入培养基。经过37℃培养16小时,观察抗霉素类似物的最小抑菌浓度(MIC)值。
对抗霉素类似物进行细胞毒性的测定,方法如下:首先将小鼠细胞(P388murine leukemia cells)在37℃于100μL RPMI 1640培养基(购于Life Technologies Corporation)中于10μg/mL青霉素和链霉素,10%牛血清,5%CO2中培养。将抗霉素类似物溶于二甲基亚砜后加入培养基中,经过3天培养后,加入50μL3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(MTT)(1mg/mL),而后继续培养4小时。经离心后,将沉淀溶于二甲基亚砜中测定其570nm的吸光度,计算半抑制浓度IC50。经过测定和计算,抗霉素类似物的抗真菌活性和细胞毒性如下表1所示。
表1
抗霉素类似物及其制备方法和用途专利购买费用说明
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
![]()















动态评分
0.0