

IPC分类号 : C08J5/18,C08G61/12,C25D9/02,C08L65/00






专利摘要
本发明公开了一种PTBTPA/PEDOT聚合物叠层复合薄膜及其制备与在电致变色材料中的应用。本发明通过叠层复合的方法,制备出颜色更丰富的电致变色聚合物薄膜。本发明成本低,操作简单,且制备过程无污染。
权利要求
1.一种PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:所述的PTBTPA/PEDOT聚合物叠层复合薄膜按照如下方法进行制备:
(1)在三电极电解池体系中,以4,4’,4”-三[4-(2-联噻吩基)苯基]胺为单体A,以1-丁基-3-甲基四氟硼酸盐为支持电解质A,以二氯甲烷和乙腈的混合溶液为电解溶剂A,所述的单体A、支持电解质A及电解溶剂A构成电解液A,以金电极、铂电极、氧化铟锡导电玻璃电极或氟掺杂氧化锡导电玻璃电极为工作电极,以金电极或铂电极为辅助电极,以银/氯化银电极为参比电极,在室温下采用恒电位法,在1.0~1.3V电压条件下,进行电化学聚合反应,当聚合电量达到0.02C~0.08C时,聚合结束,然后在-0.4~-0.8V负电压下进行脱掺杂,得到沉积在工作电极上的PTBTPA聚合物薄膜,然后淋洗、烘干;所述的单体A的初始终浓度为0.5~1mmol/L;所述的支持电解质A的初始终浓度为0.06-0.15mol/L;
(2)以3,4-乙烯二氧噻吩为单体B,1-丁基-3-甲基双(三氟甲磺酰)亚胺盐为支持电解质B,以二氯甲烷为电解溶剂B,所述的单体B、支持电解质B及电解溶剂B构成电解液B,以步骤(1)所得覆盖有PTBTPA薄膜的电极为工作电极,以金电极或铂电极为辅助电极,以银/氯化银电极为参比电极,在室温下采用恒电位法,在1.2~1.6V电压条件下进行电化学聚合反应,当聚合电量达到0.03C~0.08C时,聚合结束,然后在-0.6~-1.0V负电压下进行脱掺杂,得到沉积在工作电极上的聚合物薄膜,经淋洗、烘干,得到PTBTPA/PEDOT叠层复合聚合物薄膜;所述的单体B的初始终浓度为2~7mmol/L,所述的电解质B的初始终浓度为0.06-0.15mol/L。
2.如权利要求1所述的PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:步骤(1)中,所述的单体A的初始终浓度为0.75mmol/L。
3.如权利要求1所述的PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:步骤(1)中,所述的支持电解质A1-丁基-3-甲基四氟硼酸盐的初始终浓度为0.1mol/L。
4.如权利要求1所述的PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:步骤(1)中,所述的工作电极为氧化铟锡导电玻璃电极;所述的辅助电极为铂电极;所述的参比电极为双液接型银/氯化银电极;所述的双液接型银/氯化银电极以3mol/L的氯化钾水溶液为第一液接,以所述的电解液A为第二液接。
5.如权利要求1所述的PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:步骤(1)中,所述的恒电位法为:在1.2V电压条件下,进行电化学聚合反应,当聚合电量为0.04C,聚合结束,在-0.6V负电位下脱掺杂50~70s,得到沉积在工作电极上的PTBTPA聚合物薄膜。
6.如权利要求1所述的PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:步骤(1)中,所述的聚合物薄膜淋洗、烘干过程具体操作为:用乙腈淋洗沉积在工作电极上的PTBTPA聚合物薄膜,然后置于60~80℃真空干燥箱中干燥4~8h。
7.如权利要求1所述的PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:步骤(2)中,所述的单体B的初始终浓度为5mmol/L,所述的电解质B的初始终浓度为0.1mol/L。
8.如权利要求1所述的PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:步骤(2)中,所述的工作电极为氧化铟锡导电玻璃电极;所述的辅助电极为铂电极;所述的参比电极为双液接型银/氯化银电极;所述的双液接型银/氯化银电极以3moL/L的氯化钾水溶液为第一液接,以所述的电解液B为第二液接。
9.如权利要求1所述的PTBTPA/PEDOT聚合物叠层复合薄膜,其特征在于:步骤(2)中,所述的恒电位聚合法为:在1.4V电压条件下,当聚合电量分别为0.03C、0.05C、0.07C时,聚合结束,在-0.8V负电位下脱掺杂50~70s,分别得到沉积在工作电极上的聚合电量为0.03C的PTBTPA/PEDOT、聚合电量为0.05C的PTBTPA/PEDOT或聚合电量为0.07C的PTBTPA/PEDOT聚合物薄膜。
10.一种如权利要求1所述的PTBTPA/PEDOT叠层复合聚合物薄膜应用于制备电致变色材料。
说明书
技术领域
本发明涉及一种多色显示电致变色聚合物叠层复合薄膜(PTBTPA/PEDOT)及其制备方法与应用。
背景技术
聚合物电致变色(PEC)材料具有结构可修饰、能带可调、变色范围广、光学对比度高、加工性能好以及响应速度快等优点,在智能调光玻璃、平面显示器件、汽车自动防眩目后视镜等领域具有潜在的应用前景,被认为是下一代显示材料的发展方向之一。但PEC材料要在显示领域实现商业化应用,多色乃至全色显示性能是最亟待解决的难题之一。目前实现PEC材料多色显示性能的途径有两条:一是分子结构修饰调控,如给体-受体结构调控、多种单体共聚等。这种方式虽可实现多达5-6种颜色的显示,但难以覆盖全色范围;二是设计每层显示不同颜色的多层结构EC器件,但这无疑会增加制造工艺的复杂性,提高生产成本。
发明内容
本发明的目的在于提供一种多色显示电致变色聚合物叠层复合薄膜(PTBTPA/PEDOT)及其制备方法与应用。
本发明为解决技术问题采用如下技术方案:
一种多色显示电致变色聚合物叠层复合薄膜(PTBTPA/PEDOT)具体按如下方法进行制备:
(1)在三电极电解池体系中,以4,4’,4”-三[4-(2-联噻吩基)苯基]胺(TBTPA)为单体,以4,4’,4”-三[4-(2-联噻吩基)苯基]胺为单体A,以1-丁基-3-甲基四氟硼酸盐为支持电解质A,以二氯甲烷和乙腈的混合溶液为电解溶剂A,所述的单体A、支持电解质A及电解溶剂A构成电解液A,以金电极、铂电极、氧化铟锡导电玻璃(ITO)电极或氟掺杂氧化锡导电玻璃(FTO)电极为工作电极,以金电极或铂电极为辅助电极,以银/氯化银电极为参比电极,在室温下采用恒电位法,在1.0~1.3V电压条件下,进行电化学聚合反应,当聚合电量达到0.02C~0.08C时,聚合结束,然后在-0.4~-0.8V负电压下进行脱掺杂,得到沉积在工作电极上的PTBTPA聚合物薄膜,然后淋洗、烘干;所述的单体A的初始终浓度为0.5~1mmol/L;所述的支持电解质A的初始终浓度为0.06-0.15mol/L;
(2)以3,4-乙烯二氧噻吩为单体B,1-丁基-3-甲基双(三氟甲磺酰)亚胺盐为支持电解质B,以二氯甲烷为电解溶剂B,所述的单体B、支持电解质B及电解溶剂B构成电解液B,以步骤(1)所得覆盖有PTBTPA薄膜的电极为工作电极,以金电极或铂电极为辅助电极,以银/氯化银电极为参比电极,在室温下采用恒电位法,在1.2~1.6V电压条件下进行电化学聚合反应,当聚合电量达到0.03C~0.08C时,聚合结束,然后在-0.6~-1.0V负电压下进行脱掺杂,得到沉积在工作电极上的聚合物薄膜,经淋洗、烘干,得到PTBTPA/PEDOT叠层复合聚合物薄膜;所述的单体B的初始终浓度为2~7mmol/L,所述的电解质B的初始终浓度为0.06-0.15mol/L。
进一步,步骤(1)中,优选的,本发明中所述的4,4’,4”-三[4-(2-联噻吩基)苯基]胺(TBTPA)单体的初始终浓度为0.75mmol/L;所述的支持电解质A 1-丁基-3-甲基四氟硼酸盐([BMIM]BF4)的初始终浓度为0.1mol/L;所述的二氯甲烷和乙腈溶剂规格为色谱纯。
进一步,步骤(1)中,所述的工作电极优选为氧化铟锡导电玻璃电极;所述的辅助电极优选为铂电极;所述的参比电极优选为双液接型银/氯化银电极;所述的双液接型银/氯化银电极以3moL/L的氯化钾水溶液为第一液接,以本发明所述的电解液A为第二液接。
进一步,步骤(1)中,所述的恒电位聚合法为:在1.2V电压条件下,进行电化学聚合反应,当聚合电量为0.04C,聚合结束,在-0.6V负电位下脱掺杂50~70s,得到沉积在工作电极上的PTBTPA聚合物薄膜。
进一步,步骤(1)中,所述的聚合物薄膜淋洗、烘干过程具体操作为:用乙腈淋洗沉积在工作电极上的PTBTPA聚合物薄膜,然后置于60~80℃真空干燥箱中干燥4~8h。
进一步,步骤(2)中,所述的工作电极优选为氧化铟锡导电玻璃电极;所述的辅助电极优选为铂电极;所述的参比电极优选为双液接型银/氯化银电极;所述的双液接型银/氯化银电极以3moL/L的氯化钾水溶液为第一液接,以本发明所述的电解液B为第二液接。
进一步,步骤(2)中,所述的单体B的初始终浓度为5mmol/L;所述的电解质B的初始终浓度为0.1mol/L。
进一步,步骤(2)中,所述的恒电位聚合法为:在1.4V电压条件下,当聚合电量分别为0.03C、0.05C、0.07C时,聚合结束,在-0.8V负电位下脱掺杂50~70s,分别得到沉积在工作电极上的PTBTPA/PEDOT(0.03C)、PTBTPA/PEDOT(0.05C)或PTBTPA/PEDOT(0.07C)聚合物薄膜。
本发明所述的聚合物薄膜的光谱电化学和电致变色性能测试:通过电化学工作站与紫外一可见分光光度计联用可以对聚合物薄膜进行紫外吸收测试、对比度的测试以及响应时间的计算。通过对沉积有PTBTPA/PEDOT的工作电极施加不同电压来测试薄膜的紫外可见吸收光谱;通过双电位阶跃法来测试薄膜的动力学性能。
进一步,所述的施加不同的电压范围为-0.8~1.4V(优选为-0.8V、0.5V、0.7V、0.8V、0.9V、1.0V、1.2V、1.4V)。
进一步,所述的双电位阶跃法为:在-0.8V到1.4V之间的电致变色切换响应,电压阶跃时间为5s。
本发明通过红外光谱来表征所得聚合物薄膜的结构,证实了该聚合物薄膜的形成。
本发明所述的PTBTPA/PEDOT叠层复合聚合物薄膜应用于制备电致变色材料。
与现有技术相比,本发明的有益效果在于:
(1)可以通过叠层复合的方法,制备出颜色更丰富的电致变色聚合物薄膜。
(2)通过制备叠层复合聚合物薄膜,获得了颜色丰富、对比度较高、响应快速的电致变色薄膜。
(3)本发明成本低,操作简单,且制备过程无污染,符合绿色化学,保护环境的生态理念。
附图说明
图1是实施例1制备的PTBTPA薄膜的扫面电镜图。
图2是实施例1制备的(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜薄膜的扫面电镜图。
图3是实施例1制备的(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜的红外光谱图。
图4是实施例1制备的(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜的紫外吸收光谱图。
图5是实施例2制备的(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜的扫面电镜图。
图6是实施例2制备的(PTBTPA/PEDOT(0.05C))叠层复合聚合物薄膜在450nm波长处和1100nm波长处的光学对比度。
图7是实施例3制备的(PTBTPA/PEDOT(0.07C))叠层复合聚合物薄膜的扫面电镜图。
图8是实施例3制备的(PTBTPA/PEDOT(0.07C))叠层复合聚合物薄膜在450nm波长处和1100nm波长处的光学对比度。
具体实施方式
实施例1
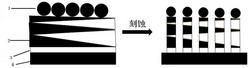
(1)在三电极电解池体系中,以TBTPA(0.05535g,0.75mmol)为单体,以[BMIM]BF4(2.26g,0.01mol)为支持电解质,CH2Cl2:ACN(70ml:30ml体积比)溶液,配制成单体浓度0.75mmol/L、支持电解质浓度0.1mol/L的混合溶液100mL为电解液,以ITO导电玻璃为工作电极,以铂电极为辅助电极,以银/氯化银电极为参比电极。在室温下采用恒电位法1.2V进行电化学聚合反应,聚合电量0.04C,然后再负电位-0.6V下脱掺杂50s,得到一层沉积在ITO工作电极上的橙色聚合物薄膜,用乙腈淋洗去除聚合物薄膜表面残留的电解液并在60℃真空干燥环境中烘干5h后得到PTBTPA薄膜。通过扫描电镜测试其表面微观形貌,如图1所示。
(2)在三电极电解池体系中,以EDOT(0.07109g,0.5mmol)为单体,以[BMIM]TF2N(4.19g,0.01mol)为支持电解质,以二氯甲烷(100mL)为电解溶剂,配制成单体浓度5mmol/L、支持电解质浓度0.1mol/L的混合溶液100mL为电解液,以上一步骤覆盖有PTBTPA薄膜的ITO玻璃为工作电极,以铂电极为辅助电极,以银/氯化银电极为参比电极。在室温下采用恒电位法1.4V进行电化学聚合反应,聚合电量0.03C,然后再负电位-0.8V下脱掺杂50s,在室温下采用恒电位法进行电化学聚合反应,得到沉积在工作电极上的聚合物薄膜,用乙腈淋洗去除聚合物薄膜表面残留的电解液并烘干后得到(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜。通过扫描电镜测试其表面微观形貌,如图2所示。
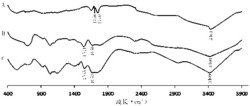
(3)(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜的红外光谱:从PTBTPA的谱图中可以看出,1602和733cm-1的峰对应于苯环中C-C伸缩振动和C-H面外振动特征峰。793和831cm-1的峰对应噻吩环C-S键的特征峰。1491和1453cm-1对应噻吩环的伸缩振动峰。1320和1270cm-1的峰对应C-N键的特征峰。从PEDOT薄膜的谱图中可以看出可知,所包含1509,1482和1301cm-1处的吸收分别属于噻吩环中C=C双键以及噻吩环中C-C单键的伸缩振动吸收。而位于1145和1059cm-1处的吸收则归因于亚乙二氧基的伸缩振动吸收。噻吩环上C-S键的振动吸收特征峰在图中位于943和840cm-1。与两者薄膜相比较,(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜的红外光谱包含了PTBTPA和PEDOT的所有特征吸收峰,说明二者成功地复合。如图3所示。
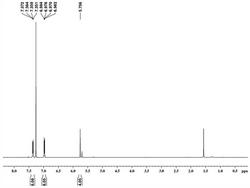
(4)(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜的光谱电化学和电致变色性能测试:通过电化学工作站与紫外一可见分光光度计联用可以对聚合物薄膜进行紫外吸收测试、对比度的测试。对步骤(1)中得到的(PTBTPA/PEDOT(0.03C))叠层复合聚合物薄膜施加-0.8V的电压时,而复合薄膜在中性态下具有两个吸收峰位于468nm最大吸收峰和594nm吸收峰分别归属于两层聚合物薄膜主链的π-π*共轭吸收;当电压逐渐增大,主链吸收强度逐渐减弱,位于800nm和大于1100nm附近极化子和双极化子的吸收不断增强。同时,可以观察到复合薄膜在-0.8V~1.4V的电压作用下呈现橙色-蓝色-墨绿色的颜色变化。根据紫外光谱图,选择了在450nm可见光区和1100nm近红外光区分别测试了该薄膜在不同波长处的光学对比度和响应时间。在450nm处对比度为25%,;在1100nm处对比度为67.8%。如图4所示。
实施例2
(1)在三电极电解池体系中,以EDOT(0.07109g,0.5mmol)为单体,以[BMIM]TF2N(4.19g,0.01mol)为支持电解质,以二氯甲烷(100mL)为电解溶剂,配制成单体浓度5mmol/L、支持电解质浓度0.1mol/L的混合溶液100mL为电解液,以上一步骤覆盖有PTBTPA薄膜的ITO玻璃为工作电极,以铂电极为辅助电极,以银/氯化银电极为参比电极。在室温下采用恒电位法1.4V进行电化学聚合反应,聚合电量0.05C,然后再负电位-0.8V下脱掺杂50s,在室温下采用恒电位法进行电化学聚合反应,得到沉积在工作电极上的聚合物薄膜,用乙腈淋洗去除聚合物薄膜表面残留的电解液并烘干后得到(PTBTPA/PEDOT(0.05C))叠层复合聚合物薄膜。通过扫描电镜测试其表面微观形貌,如图5所示。
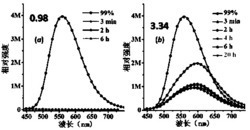
(2)(PTBTPA/PEDOT(0.05C))叠层复合聚合物薄膜的光谱电化学和电致变色性能测试:通过电化学工作站与紫外一可见分光光度计联用可以对聚合物薄膜进行紫外吸收测试、对比度的测试。根据紫外光谱图,选择了在450nm可见光区和1100nm近红外光区分别测试了该薄膜在不同波长处的光学对比度和响应时间。在450nm处对比度为18%,;在1100nm处对比度为45%。如图6所示。
实施例3
(1)在三电极电解池体系中,以EDOT(0.07109g,0.5mmol)为单体,以[BMIM]TF2N(4.19g,0.01mol)为支持电解质,以二氯甲烷(100mL)为电解溶剂,配制成单体浓度5mmol/L、支持电解质浓度0.1mol/L的混合溶液100mL为电解液,以上一步骤覆盖有PTBTPA薄膜的ITO玻璃为工作电极,以铂电极为辅助电极,以银/氯化银电极为参比电极。在室温下采用恒电位法1.4V进行电化学聚合反应,聚合电量0.07C,然后再负电位-0.8V下脱掺杂50s,在室温下采用恒电位法进行电化学聚合反应,得到沉积在工作电极上的聚合物薄膜,用乙腈淋洗去除聚合物薄膜表面残留的电解液并烘干后得到(PTBTPA/PEDOT(0.07C))叠层复合聚合物薄膜。通过扫描电镜测试其表面微观形貌,如图7所示。
(2)(PTBTPA/PEDOT(0.07C))叠层复合聚合物薄膜的光谱电化学和电致变色性能测试:通过电化学工作站与紫外一可见分光光度计联用可以对聚合物薄膜进行紫外吸收测试、对比度的测试。根据紫外光谱图,选择了在450nm可见光区和1100nm近红外光区分别测试了该薄膜在不同波长处的光学对比度和响应时间。在450nm处对比度为5%,;在1100nm处对比度为42%。如图8所示。
一种PTBTPA/PEDOT聚合物叠层复合薄膜及其制备与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0