







专利摘要
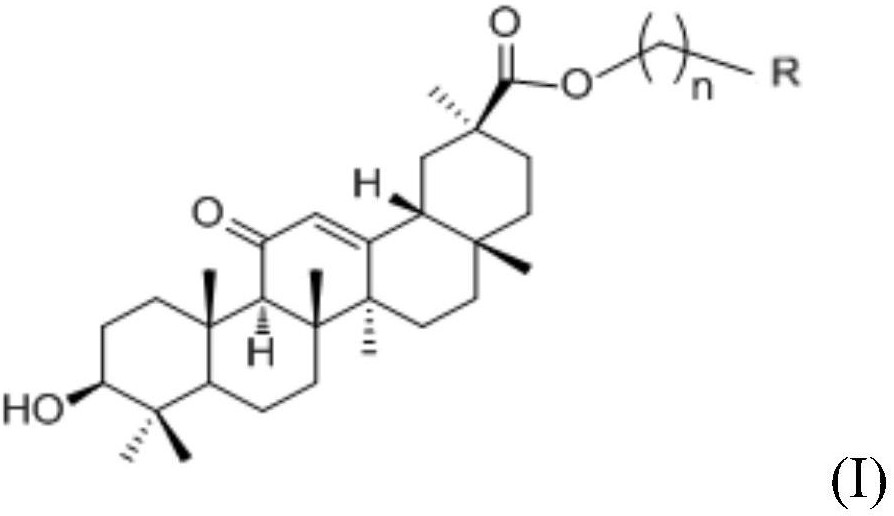
本发明公开了一种羊毛甾烷型三萜类化合物及其制备方法和用途,属于医药领域。本发明结构式为本发明化合物的分离方法是将毛果南烛叶的氯仿部位由正相硅胶柱划段,以CH2Cl2‑MeOH(50:1~0:1)进行洗脱,将得到的部分再进行正相CH2Cl2‑MeOH(40:1~0:1)、反相硅胶以MeOH‑H2O(20:1,40:1,60:1,80:1)洗脱划段,再经凝胶、HPLC进行分离纯化。本发明化合物可用于制备抗肿瘤药物或作为抗肿瘤药物开发的先导化合物。
权利要求
1.一种羊毛甾烷型三萜类化合物,其特征在于所述的羊毛甾烷型三萜类化合物为 结构式中R1为β-D-Glc、R2为CO2H,或者此结构式中R1为β-D-Glc-(1→2)-α-L-Rha、R2为CH2OH,或者此结构式中R1为β-D-Glc-(1→2)-α-L-Rha、R2为CHO,或者此结构式中R1为β-D-Glc-(1→2)-α-L-Rha、R2为CH3。
2.如权利要求1所述的一种羊毛甾烷型三萜类化合物的制备方法,其特征在于所述的制备方法是按下述步骤进行的:
步骤一、将毛果南烛叶干燥后粉碎,用95%(体积)乙醇提取3次,减压浓缩得到总浸膏;
步骤二、总浸膏加适量蒸馏水混悬,依次用石油醚、氯仿、乙酸乙酯和正丁醇萃取;
步骤三、步骤二萃取物的氯仿部位用硅胶按1:1的质量比拌样,用CH2Cl2-MeOH进行梯度(50:1→30:1→20:1→15:1→12:1→8:1→5:1→3:1→0:1)洗脱;
步骤四、将步骤三中体积比为12:1的CH2Cl2-MeOH洗脱部位过正向硅胶柱划段,再用CH2Cl2-MeOH进行梯度(40:1→30:1→20:1→15:1→10:1→5:1→0:1)洗脱,取体积比为(15~0):1的CH2Cl2-MeOH洗脱部位,再经中压ODS柱子,依次用20%(体积)MeOH-H2O、40%MeOH-H2O、60%MeOH-H2O、80%MeOH-H2O洗脱,将80%甲醇-水洗脱部位经Sephadex LH-20柱子用纯MeOH洗脱,每20mL作为一个单位接收,经TLC检测,合并相同样点接受液,共分为14个部分,浓缩至干,其中第六部分再经半制备HPLC(戴安P680高效液相色谱仪,UVD170紫外检测器,RPC-18柱子(250×10nm,Welch))进一步分离,以流动相MeCN-H2O(67:33,v/v,流速:1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L-鼠李糖]-羊毛甾-25,30-二醇-24-羰基-8-烯(2.6mg,tR=19.6min);其中第七部分再经半制备HPLC(戴安P680高效液相色谱仪,UVD170紫外检测器,RP C-18柱子(250×10nm,Welch)进一步分离,以流动相MeCN-H2O(80:20,v/v,流速:1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L-鼠李糖]-羊毛甾-25-羟基-24-羰基-30-醛-8-烯(6.4mg,tR=24.3min)和化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L-鼠李糖]-羊毛甾-25-羟基-24-羰基-8-烯(8.8mg,tR=38.2min);
按步骤三将体积比为20:1(70g)的CH2Cl2-MeOH洗脱部位,过聚酰胺柱子(除掉黄酮类物质)用蒸馏水洗脱,合并所得水部位(35g),再过正向硅胶柱划段,以CH2Cl2-MeOH(30:1→20:1→15:1→4:1)进行洗脱,再经中压ODS柱子,以MeOH-H2O(20:1,40:1,60:1,80:1)洗脱,其中80%MeOH(20.4g)洗脱产品,经TLC检测,合并相同样点接受液,共7个部分,浓缩至干,将其中第五部分再经凝胶Sephadex LH-20分离,得到的粗品(48mg),每20mL作为一个单位接收经HPLC纯化,以流动相MeCN-H2O-CH3COOH(80:20:0.1,v/v/v,流速1.5mL/min)洗脱,检测波长设定为210nm,每次进样量为80μL,得到3-ɑ-β-D-葡萄糖-羊毛甾-25-羟基-24-羰基-8-烯-30-酸(4mg,tR=26.8min)。
3.根据权利要求2所述一种羊毛甾烷型三萜类化合物的制备方法,其特征在于步骤一中用95%乙醇提取的时间间隔为24h,温度为45℃,固液比为1:3。
4.根据权利要求2所述一种羊毛甾烷型三萜类化合物的制备方法,其特征在于将步骤三中将步骤二得到的氯仿提取物用100~200目硅胶按照质量1:1拌样,依次以CH2Cl和MeOH的体积比为50:1→45:1→35:1→25:1→15:1→10:1→5:1→1:1→0:1进行梯度洗脱。
5.如权利要求1所述一种羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗肿瘤药物中的应用。
6.根据权利要求5所述一种羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备治疗白血病药物中的应用。
7.根据权利要求5所述一种羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗乳腺癌药物中的应用。
8.根据权利要求5所述一种羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗肝癌药物中的应用。
9.根据权利要求5所述一种羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗结肠癌药物中的应用。
10.根据权利要求5所述一种羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗肺癌药物中的应用。
说明书
技术领域:
本发明属于医药技术领域;具体涉及一种羊毛甾烷型三萜类化合物及其制备方法和用途。
背景技术:
恶性肿瘤(malignant tumor)又称癌症,特征为局部组织的细胞基因表达调控异常而失去了对细胞生长的正常调控,并逃避了免疫系统的监视,引起变异细胞过度增生并由原发部位向它处播散,这种播散如无法控制,将侵犯要害器官并引起衰竭,最后导致死亡。为此,癌症的防治刻不容缓,这已成为全世界重点关注的课题。国际上临床常用的抗肿瘤药植物药和辅助化疗的中药主要包括紫杉醇、喜树碱、长春新碱、人参等,它们在肿瘤治疗中疗效确切、不良反应小,成为抗肿瘤药物市场上的主角。我国可药用植物资源丰富,从传统中草药中挖掘抗肿瘤活性成分,具有明显的优势,尤其是三萜皂苷类化合物近年来广受科学工作者的青睐。近期研究表明三萜皂苷通过诱导细胞凋亡和分化的作用机制在抗恶性肿瘤方面发挥出抗扩散、抗代谢、抗血管生成、逆转耐药等多种功能,还可以降低放疗和化疗的副作用,因此三萜皂苷在抗恶性肿瘤药物的研究领域具有显著的意义。
毛果南烛(Lyonia ovalifolia var.hebecarpa),也称为毛果珍珠花、毛果米饭花,是珍珠花(L.ovalifolia)的一个变种,生于海拔1400~3400米的阳坡灌丛中,主要产于江苏、安徽、浙江、广东、广西、四川、云南西北部等,茎、叶可药用,有强筋益气、收敛止泻效用。以往植物学家对其化学成分的研究主要集中在结构多样的二萜类化合物上,对三萜类化合物的研究并不十分全面,而羊毛甾烷型三萜多分布于海参属和灵芝属中,在植物中极为罕见,仅在百合科,风信子科,马鞭草科,豆科和杜鹃花科植物中被发现过,且具有多种生物活性,具有潜在的药用价值。
发明内容:
本发明的目的是提供一种羊毛甾烷型三萜类化合物及其制备方法和用途。
本发明从植物毛果南烛叶中分离提取的多种羊毛甾烷型三萜类化合物,提供了羊毛甾烷型三萜类化合物的制备方法。
以顺铂(Cis-platinum MW 300)为阳性对照,本发明所述的三萜类化合物具有一定的抑制肿瘤细胞增殖活性,包括HL-60(人早幼粒白血病细胞)、MCF-7(乳腺癌细胞)、SMMC-7221(人肝癌细胞)、A-549(人肺腺癌细胞)、SW480(人结肠癌细胞)和BEAS-2B(肺上皮细胞)。这些化合物可用于制备抗肿瘤药物或作为抗肿瘤药物开发的先导化合物。
本发明的化合物结构式:
,其中化合物1-4中
本发明中羊毛甾烷型三萜类化合物的制备方法是按下述步骤进行的:
步骤一、将毛果南烛叶干燥后粉碎,用95%(体积)乙醇提取3次,减压浓缩得到总浸膏;
步骤二、总浸膏加适量蒸馏水混悬,依次用石油醚、氯仿、乙酸乙酯和正丁醇萃取;
步骤三、步骤二萃取物的氯仿部位用硅胶按一定质量比(1:1)拌样,用 CH2Cl2-MeOH进行梯度(50:1→30:1→20:1→15:1→12:1→8:1→5:1→3:1→0:1)洗脱;步骤四、将步骤三中体积比为15:1的CH2Cl2-MeOH洗脱部位过正向硅胶柱划段,再用CH2Cl2-MeOH进行梯度(40:1→30:1→20:1→15:1→10:1→5:1→0:1)洗脱,取体积比为(15~0):1的CH2Cl2-MeOH洗脱部位,再经中压ODS(填料为YMC GEL ODS-AS 50μm,Japan)柱子,依次用20%(体积)MeOH-H2O、40%MeOH-H2O、 60%MeOH-H2O、80%MeOH-H2O洗脱,将80%甲醇-水洗脱部位经Sephadex LH-20 柱子用纯MeOH洗脱,每20mL作为一个单位接收,经TLC检测,合并向同样点接受液,共分为14个部分,浓缩至干,其中第六部分再经半制备HPLC(戴安P680高效液相色谱仪,UVD170紫外检测器,RP C-18柱子(250×10nm,Welch))进一步分离,以流动相MeCN-H2O(67:33v/v,流速:1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L-鼠李糖]-羊毛甾-25,30- 二醇-24-羰基-8-烯(2.6mg,tR=19.6min);其中第七部分再经半制备HPLC(戴安P680 高效液相色谱仪,UVD170紫外检测器,RP C-18柱子(250×10nm,Welch))进一步分离,以流动相MeCN-H2O(80:20,v/v,流速:1.5mL/min),检测波长设定为210nm, 每次进样量为80μL,得到化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L-鼠李糖]-羊毛甾-25-羟基-24-羰基-30-醛-8-烯(6.4mg,tR=24.3min)和化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L- 鼠李糖]-羊毛甾-25-羟基-24-羰基-8-烯(8.8mg,tR=38.2min);
按步骤三将体积比为20:1(70g)的CH2Cl2-MeOH洗脱部位,过聚酰胺柱子(除掉黄酮类物质)用蒸馏水洗脱,合并所得水部位(35g),再过正向硅胶柱划段,以 CH2Cl2-MeOH(30:1→20:1→15:1→4:1)进行洗脱,再经中压ODS柱子,以MeOH-H2O (20:1,40:1,60:1,80:1)洗脱,其中80%MeOH(20.4g)洗脱产品,经TLC检测,合并相同样点接受液,共7个部分,浓缩至干,将其中第五部分再经凝胶Sephadex LH-20分离,得到的粗品(48mg),每20mL作为一个单位接收经HPLC纯化,以流动相MeCN-H2O-CH3COOH(80:20:0.1,v/v/v,流速1.5mL/min)洗脱,检测波长设定为210nm,每次进样量为80μL,得到3-ɑ-β-D-葡萄糖-羊毛甾-25-羟基-24-羰基-8-烯-30- 酸(4mg,tR=26.8min)。
上述羊毛甾烷型三萜类化合物在制备抗肿瘤药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备治疗白血病药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备抗乳腺癌药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备抗肝癌药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备抗结肠癌药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备抗肺癌药物中的应用。
本发明的羊毛甾烷型三萜具有6/6/6/5四环结构,通常含有8个甲基取代(C-4 二甲基取代,C-10,C-13,C-14,C-20和C-25二甲基取代),天然植物中此类化合物结构稳定,新化合物较少见,本发明中的这类羊毛甾烷型三萜的C-3位上单糖或二糖取代且C-24羰基取代,同时C-14位被氧化为醇羟基或羧基,极为罕见。
本发明首次从杜鹃花科植物L.ovalifolia var.hebecarpa.中发现羊毛甾烷型三萜类化合物并提供了这类新化合物的提取分离技术、结构鉴定方法和抗肿瘤细胞增殖方面的用途。
附图说明:
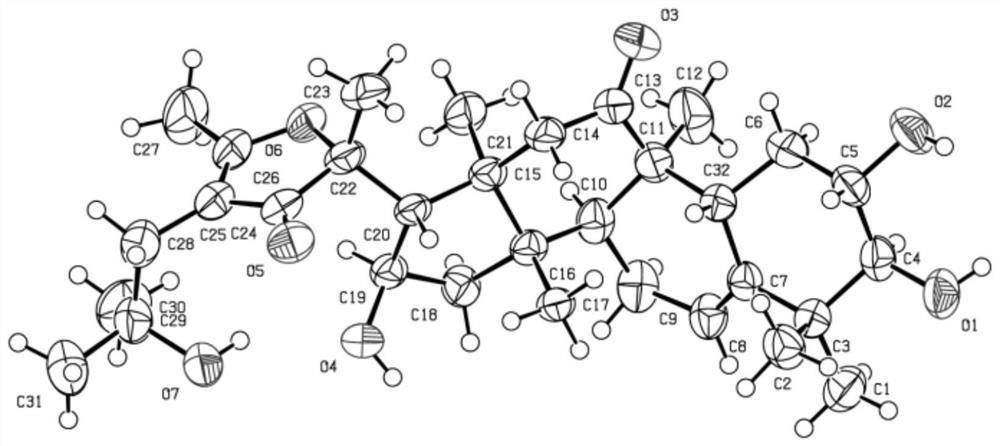
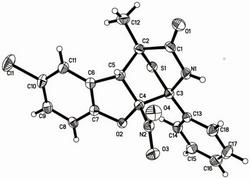
图1是化合物1的1HNMR图谱(in CD3OD 400MHz);图2是化合物1的13C NMR 图谱(inCD3OD 100MHz);图3是化合物1的DEPT图谱(in CD3OD 100MHz);图4 是化合物1的HSQC图谱(in CD3OD 400MHz);图5是化合物1的1H-1H COSY图谱(in CD3OD 400MHz);图6是化合物1的HMBC图谱(in CD3OD 400MHz);图7 是化合物1的NOESY图谱(in CD3OD 400MHz);图8是化合物1的高分辨率质谱;图9是化合物2的1H NMR图谱(inPyridine-d5400MHz);图10是化合物2的13C NMR 图谱(inPyridine-d5100MHz);图11是化合物2的DEPT图谱(1inPyridine-d500MHz);图12是化合物2的HSQC图谱(inPyridine-d5400MHz);图13是化合物2的1H-1HCOSY图谱(400MHz);图14是化合物2的HMBC图谱(400MHz);图15是化合物2的NOESY图谱(inPyridine-d5400MHz);图16是化合物2的高分辨率质谱;图17 是化合物1~41-1H-1HCOSY和关键HMBC相关图。
具体实施方式
实施例1:本实施例中羊毛甾烷型三萜类化合物制备方法是按下述步骤进行的:
步骤一、干燥毛果南烛叶50kg经粉碎,用95%乙醇(24h/次,220L,45C)提取3 次,减压浓缩得到总浸膏9.15kg。
步骤二、将总浸膏加9.15kg蒸馏水混悬,依次用石油醚、氯仿、乙酸乙酯和正丁醇萃取,氯仿部位得到萃取物5.09kg。
步骤三、步骤二萃取物的氯仿部位用100~200目硅胶按照质量1:1拌样,以CH2Cl2-MeOH进行梯度(50:1→30:1→20:1→15:1→12:1→8:1→5:1→3:1→0:1)洗脱,得到9个部分。
步骤四、化合物2,3,4的制备:取步骤三体积比为12:1的CH2Cl2-MeOH洗脱部位(43.5g),过聚酰胺柱子(除掉黄酮类物质)合并水部位(33g),过正向硅胶柱划段,以CH2Cl2-MeOH(40:1→30:1→20:1→15:1→10:1→5:1→0:1)进行梯度洗脱,再经ODS(填料为YMCGEL ODS-AS 50μm,Japan)柱子,依次用20%(体积) MeOH-H2O、40%MeOH-H2O、60%MeOH-H2O、80%MeOH-H2O洗脱,其中将80%甲醇-水洗脱部位(20.0g)经Sephadex LH-20柱子用纯MeOH洗脱,每20mL作为一个单位接收,经TLC检测,合并相同样点接受液,共分为14个部分,浓缩至干,其中第六部分粗品(142mg)再经半制备HPLC(戴安P680高效液相色谱仪,UVD170紫外检测器,RP C-18柱子(250×10nm,Welch))进一步分离,以流动相MeCN-H2O (67:33,v/v,流速:1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L-鼠李糖]-羊毛甾-25,30-二醇-24-羰基-8-烯(2.6mg, tR=19.6min);其中第七部分再经半制备HPLC(戴安P680高效液相色谱仪,UVD170 紫外检测器,RP C-18柱子(250×10nm,Welch))进一步分离,以流动相MeCN-H2O (80:20,v/v,流速:1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L-鼠李糖]-羊毛甾-25-羟基-24-羰基-30-醛-8-烯(6.4 mg,tR=24.3min)和化合物3ɑ-O-[β-D-葡萄糖-(2→1)-ɑ-L-鼠李糖]-羊毛甾-25-羟基-24- 羰基-8-烯(8.8mg,tR=38.2min);
化合物1的制备:将步骤三体积比为20:1(70g)的CH2Cl2-MeOH洗脱部位,过聚酰胺柱子(除掉黄酮类物质)用蒸馏水洗脱,合并所得水部位(35g),再过正向硅胶柱划段,以CH2Cl2-MeOH(30:1→20:1→15:1→4:1)进行洗脱,再经中压ODS 柱子,以MeOH-H2O(20:1,40:1,60:1,80:1)洗脱,其中80%MeOH(20.4g)洗脱产品,经TLC检测,合并相同样点接受液,共7个部分,浓缩至干,将其中第五部分再经凝胶Sephadex LH-20分离,得到的粗品(48mg),每20mL作为一个单位接收经HPLC纯化,以流动相MeCN-H2O-CH3COOH(80:20:0.1,v/v/v,流速1.5mL/min) 洗脱,检测波长设定为210nm,每次进样量为80μL,得到3-ɑ-β-D-葡萄糖-羊毛甾-25- 羟基-24-羰基-8-烯-30-酸(4mg,tR=26.8min)。
通过NMR、化学反应方法确定化合物1,2,3,4的绝对构型。
化合物1:白色粉末;-23(c 0.2,MeOH);1H NMR and 13C NMR图谱数据见表1 和2;HRESIMS m/z 673.3913[M+Na]+(calcd for C36H58O10Na,673.3928).
化合物2:白色粉末;-82(c 0.2,MeOH);1HNMR and 13C NMR图谱数据见表1 和2;HRESIMS m/z 805.4705[M+Na]+(calcd for C42H70O13Na,805.4714).
化合物3:白色粉末;-188(c 0.6,MeOH);1HNMR and 13C NMR图谱数据见表1 和2;HRESIMS m/z 803.4526[M+Na]+(calcd for C42H68O13Na,803.4558).
化合物4:白色粉末;-27(c 0.4,MeOH);1HNMR and 13C NMR图谱数据见表1 和2;HRESIMS m/z 789.4764[M+Na]+(calcd for C42H70O12Na,789.4765).
本发明的化合物1~4结构相似,仅在C-3和C-14上取代基存在差异,图1-16提供化合物1和2的1H NMR、13C NMR、DEPT、HSQC、HMBC、1H-1H COSY、NOESY、 HRESIMS,并提供了化合物1~4的1H-1H COSY和关键HMBC相关图见图17
化合物1,2,3,4的1H NMR和13C NMR数据见表1和2.
Table 1.1H NMR[δ,mult,(Jin Hz)]Data for Compounds 1~4(400MHz)
aRecorded in methanol-d4.bRecorded in pyridine-d5.
Table 2.13C NMR Data for Compounds 1-4(100 MHz)
aRecorded in methanol-d4.bRecorded inpyridine-d
化合物的糖苷键的酸水解
为了确定化合物1中葡萄糖的绝对构型,将化合物1的酸水解:将化合物1(6mg) 溶于2mL甲醇中,加入2mol/L的HCl水溶液2mL,70℃下水解96h,反应液减压蒸干至中性,加入1mL蒸馏水混匀,再用1mL乙酸乙酯萃取3次。水层减压浓缩蒸干得到粗单糖(0.5mg)。
单糖的衍生化:向水解得到的单糖(0.5mg)、D-(1mg)和L-葡萄糖标准品(1mg) 中分别加入2mg的L-半胱氨酸甲酯盐酸盐,0.2mL的无水吡啶,65℃下反应2h,随后在混合液中加入0.2mLN-三甲基硅咪唑65℃下继续反应1.5h,减压蒸干,加入 1mL蒸馏水,0.2mL正己烷萃取,有机层用GC检测。
GC检测条件:WM-1毛细管柱(30m*0.25mm*0.5μm,Welch);柱温:220℃保留 5min,每分钟5℃升温到270℃,进样口温度:250℃,载气:N2,1.0mL/min,分流比:10:1;FID检测器,检测温度:250℃。
化合物1中糖衍生物经GC分析,保留时间为13.31min,D-和L-葡萄糖标准品的衍生化产物的保留时间分别为13.35和13.85min,因此,化合物1的糖为D-葡萄糖。
化合物2的水解:将化合物2(8mg)溶于2mL甲醇中分四瓶,各加入2mol/L 的HCl水溶液2mL,70℃下水解96h,反应液减压蒸干至中性,加入1mL蒸馏水混匀,再用1mL乙酸乙酯萃取3次。水层减压浓缩蒸干得到粗单糖(0.8mg)。
单糖的衍生化:向水解得到的单糖(0.8mg)、D-(1mg)和L-葡萄糖、D-(1mg)和 L-鼠李糖标准品(1mg)中分别加入2mg的L-半胱氨酸甲酯盐酸盐,0.2mL的无水吡啶,65℃下反应2h,随后在混合液中加入0.2mLN-三甲基硅咪唑65℃下继续反应 1.5h,减压蒸干,加入1mL蒸馏水,0.2mL正己烷萃取,有机层用GC检测。
GC检测条件:WM-1毛细管柱(30m*0.25mm*0.5μm,Welch);柱温:220℃保留 5min,每分钟5℃升温到270℃,进样口温度:250℃,载气:N2,1.0mL/min,分流比:10:1;FID检测器,检测温度:250℃。
化合物2中糖衍生物经GC分析,保留时间为13.33和13.67min,D-、L-葡萄糖和L-鼠李糖标准品的衍生化产物的保留时间分别为13.35、13.85和13.56min,因此,化合物2的糖部分位为D-葡萄糖和L-鼠李糖。
化合物3,4采用相同的化学方法确定的糖部分的绝对构型,以D、L-葡萄糖和L- 鼠李糖标准品硅烷化衍生物为对照,经GC检测保留时间分别为13.34和13.67,13.33 和13.64min,故化合物3和4中糖的绝对构型均为D-葡萄糖和L-鼠李糖。
实施例2化合物1,2,3,4对HL-60(人早幼粒白血病细胞)、MCF-7(乳腺癌细胞)、SMMC-7221(人肝癌细胞)、A-549(人肺腺癌细胞)、SW480(人结肠癌细胞) 和BEAS-2B(肺上皮细胞)五种肿瘤细胞增殖的抑制活性,这四种化合物对正常细胞均无毒副作用(IC50>40μM)。且其中化合物4具有显著抑制HL-60和SMMC-7721 体外增殖活性,具体数据见表3
Table 3.Anti-proliferativeActivities ofCompounds 1-4against FiveCancer Cell Lines and One Normal cell line(IC50inμM)a
a Other compounds:IC50>40μM.b cis-Platin was used as apositivecontrol.
实验结论:羊毛甾烷型三萜单糖苷及二糖苷,具有不同程度抑制肿瘤细胞增殖活性,其C-3位的糖部分取代基团差异和C-14位的取代基差异对该类化合物抗肿瘤活性的强弱程度及选择性有较大影响。
利用MTS法,研究这4个新化合物对HL-60(人早幼粒白血病细胞)、MCF-7 (乳腺癌细胞)、SMMC-7721(人肝癌细胞)、A-549(人肺腺癌细胞)、SW480(人结肠癌细胞)和BEAS-2B(肺上皮细胞)五种肿瘤细胞增殖抑制活性,本发明发现化合物4有抑制HL-60,SMMC-7721两种肿瘤细胞增殖的活性。
本发明筛查了化合物1,2,3,4对HL-60,A-549,SMMC-7721,MCF-7,SW480 五种肿瘤细胞增殖的抑制活性。化合物1,2,3,4均对正常细胞BEAS-2B无毒副作用,表示其具有显著的抗肿瘤活性。
一种羊毛甾烷型三萜类化合物及其制备方法和用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0