

IPC分类号 : B01J31/02,C07B53/00,C07C231/10,C07C237/22,C07D263/42,C07D401/04







专利摘要
本发明公开了3,4‑二氨基吡啶氮氧类手性催化剂及其在Steglich重排中的应用,属于有机化学中不对称合成技术领域。以3‑溴4‑硝基吡啶氮氧和D或L‑脯氨酰胺为原料,经过氯化和氨化后得到手性3,4‑二氨基吡啶氮氧催化剂,该催化剂用于O‑酰基二氢唑酮不对称Steglich重排反应,得到α‑季碳手性中心C‑酰基二氢吡唑酮,取得了高收率和优异的对映选择性。本发明中催化剂结构新型,合成方法简单,催化重排效果优异。
权利要求
1.3,4-二氨基吡啶氮氧类手性催化剂,其特征在于,结构为 或其对映异构体;其中,R1为C1-C8烷基,R1...R1为C2-C6环烷基,R2为C1-C8烷基、芳基或取代芳基。
2.根据权利要求1所述的手性催化剂,其特征在于,结构为: 或其对映异构体;其中,R2为芳基或C1-C6取代芳基。
3.根据权利要求1或2所述的手性催化剂,其特征在于,结构如下:
4.3,4-二氨基吡啶氮氧类手性催化剂在不对称Steglich重排反应中的应用,其特征在于,反应方程式为:
包括如下步骤:将O-酰基二氢吡唑酮和手性3,4-二氨基吡啶氮氧手性催化剂在有机溶剂中反应,得到产物C-酰基二氢唑酮。
5.根据权利要求4中的应用,其特征在于:R1选自:甲基、乙基、正丙基、正丁基、-CH2-CH2-、-CH2-CH2-CH2-;R2选自:芳基和烷基,包括:苯基、 甲基、乙基、正丙基、异丙基、叔丁基、环戊基、环己基、1-金刚烷基、2-金刚烷基、苄基、 R3选自:苯基、苄基、卤代烃基;R4选自:甲基、乙基、正丙基、异丙基、叔丁基、苄基、烷硫基、烯丙基。
6.根据权利要求4中的应用,其特征在于:所述有机溶剂为二氯甲烷、四氢呋喃、甲苯、二甲苯或二噁烷。
7.根据权利要求4中的应用,其特征在于:所述3,4-二氨基吡啶氮氧手性催化剂与O-酰基二氢吡唑酮摩尔比为1:0.01-0.1;反应温度为-40℃至25℃。
8.3,4-二氨基吡啶氮氧类手性催化剂的制备方法,其特征在于,包括如下步骤,反应方程式如下:
1)
2)
9.根据权利要求8所述的制备方法,其特征在于:所述Base选自碳酸钾、碳酸钠、碳酸铯、三乙胺、二异丙基乙基胺。
10.如权利要求1或2中所述催化剂在制备3,4-二氨基吡啶类手性催化剂中的应用,其特征在于:3,4-二氨基吡啶氮氧类手性催化剂,在还原剂存在下,反应后得到3,4-二氨基吡啶类手性催化剂,反应方程式如下:
说明书
技术领域
本发明涉及一种新型手性催化剂以及在不对称反应中的应用,具体涉及3,4-二氨基吡啶氮氧类手性催化剂及其在Steglich重排中的应用,属于有机化学中的不对称合成技术领域。
背景技术
酰基转移是自然界和有机合成中最常用的转化之一。在过去的二十年中,手性非酶促酰基转移催化剂的开发已成为一个充满活力的研究领域,已报道结构多样的催化剂((a)Ruble,J.C.;Fu,G.C.J.Am.Chem.Soc.1998,120,11532.(b)Shaw,S.A.;Aleman,P.;Vedejs,E.J.Am.Chem.Soc.2003,125,13368.(c)Joannesse,C.;Johnston,C.P.;Smith,A.D.Angew.Chem.,Int.Ed.2009,48,8914.(d)Zhang,Z.;Xie,F.;Jia,J.;Zhang,W.J.Am.Chem.Soc.2010,132,15939.(e)Uraguchi,D.;Ooi,T.Angew.Chem.,Int.Ed.2010,49,5567.(f)Cruchter,T.;Meggers,E.ACS Catal.2017,7,5151.),这些酰基转移催化剂中,手性4-(二甲氨基)自Vedejs和Fu的开创性研究以来,吡啶(DMAP)和密切相关的4-吡咯烷基吡啶(PPY)((a)Vedejs,E.;Chen,X.J.Am.Chem.Soc.1996,118,1809.(b)Kawabata,T.;Fuji,K.J.Am.Chem.Soc.1997,119,3169.(c)Ruble,J.C.;Fu,G.C.J.Am.Chem.Soc.1998,120,11532.(d)Crittall,M.R.;Rzepa,H.S.;Carbery,D.R.Org.Lett.2011,13,1250.(e)Mandai,H.;Fujii,K.;Suga,S.Nat.Commun.2016,7,11297.)一直是特别有吸引力的目标,这些DMAP类催化剂利用吡啶环上N作为亲核位点,利用吡啶氮氧上氧原子作为亲核位点至今还未有人报道。
在有机合成中,季碳手性中心的建立一直是一个具有挑战性的课题,利用手性亲核催化剂条件下发生酰基转移的Steglich重排是少数几个能获得上述产物的方法之一。
但是该现有技术在实际使用过程中仍存在各种缺陷,主要表现在在催化剂合成方面需要用到昂贵的试剂以及需要经过多步转化,催化应用中对映选择性欠佳以及催化剂活性低。所以,需要寻找更加有效的亲核催化剂以进一步解决上述问题。
发明内容
为了克服上述技术缺陷,本发明提供了一种结构新型的3,4-二氨基吡啶氮氧类手性催化剂。以3-溴4-硝基吡啶氮氧和D或L-脯氨酰胺为原料,经过氯化和氨化后得到手性3,4-二氨基吡啶氮氧催化剂,该催化剂用于O-酰基二氢唑酮不对称Steglich重排反应,得到α-季碳手性中心C-酰基二氢吡唑酮,取得了高收率和优异的对映选择性。
本发明是通过以下技术方案实现:3,4-二氨基吡啶氮氧类手性催化剂,结构为:
其中,R1为C1-C8烷基,R1...R1为C2-C6环烷基,R2为C1-C8烷基、芳基或取代芳基。
进一步优选结构为:
其中,R2为芳基或C1-C6取代芳基。
上述结构催化剂可以用于手性C-酰基二氢吡唑酮的制备,其特征在于:将O-酰基二氢吡唑酮和手性3,4-二氨基吡啶氮氧氧催化剂在有机溶剂中混合后进行不对称反应,得到产物C-酰基二氢唑酮。
反应方程式如下:
其中,R1选自:甲基、乙基、正丙基、正丁基、-CH2-CH2-、-CH2-CH2-CH2-;R2选自:各种芳基和烷基,包括:苯基、 甲基、乙基、正丙基、异丙基、叔丁基、环戊基、环己基、1-金刚烷基、2-金刚烷基、苄基、 R3选自:苯基、苄基、卤代烃基;R4选自:甲基、乙基、正丙基、异丙基、叔丁基、苄基、烷硫基、烯丙基。
进一步地,在上述技术方案中,所述有机溶剂为二氯甲烷、四氢呋喃、甲苯、二甲苯、二噁烷。
进一步地,在上述技术方案中,所述3,4-二氨基吡啶氮氧催化剂与O-酰基二氢吡唑酮摩尔比为1:0.01-0.2。
进一步地,在上述技术方案中,所述反应温度为-40℃至25℃,反应时间6-40小时。
手性3,4-二氨基吡啶氮氧类催化剂的制备,包括如下步骤:以3-溴-4-硝基吡啶氮氧和L-脯氨酰胺为原料,经过氯化和氨化后得到手性3,4-二氨基吡啶氮氧类催化剂,反应方程式如下:
进一步地,在上述合成中,碱选自:碳酸钾、碳酸钠、碳酸铯、三乙胺、二异丙基乙基胺。
进一步地,在上述合成中,所述有机溶剂选自:四氢呋喃、甲苯、1,2二氯乙烷、乙腈。
进一步地,在上述合成中,反应温度选自50-160℃。优选第一步与L-脯氨酰胺反应温度为50-120℃,第二步与乙酰氯反应温度为90-110℃,第三步与二烷基胺反应温度为80-160℃。
在合成过程中,调整反应顺序,例如:从3-溴-4-二取代氨基吡啶氮氧和L-脯氨酰胺在碱(碳酸钾、碳酸铯、三乙胺)和催化剂{三氟甲烷磺酸铜、醋酸钯、三(二亚苄基丙酮)二钯}下,在溶剂(四氢呋喃、1,2-二氯乙烷、甲苯、乙腈)中加热直接取代得不到产物。
以4位为四氢吡咯为例:
尝试采用3-溴-4-二取代氨基吡啶与L-脯氨酰胺在碱(碳酸钾、碳酸铯、三乙胺)和催化剂{三氟甲烷磺酸铜、醋酸钯、三(二亚苄基丙酮)二钯}下在溶剂(四氢呋喃、1,2-二氯乙烷、甲苯、乙腈)中加热先进行取代,随后再氧化的方式合成目标催化剂3,4-二氨基吡啶氮氧类催化剂。然而在第一步取代时就遇到困难,无法获得产物。
以4位为四氢吡咯为例:
为了对比技术效果,3,4-二氨基吡啶类催化剂也进行了合成,将上述得到的3,4-二氨基吡啶氮氧类催化剂接着与还原剂反应,一步即可得到3,4-二氨基吡啶类催化剂。反应方程式如下:
进一步地,在上述合成中,所述有机溶剂选自:四氢呋喃、甲苯、1,2-二氯乙烷。
进一步地,在上述合成中,所述反应温度为:50-160℃。优选70-110℃。
进一步地,在上述合成中,所述还原剂为:Pd/C-H2、Pt/C-H2、铁粉/盐酸、锌粉/盐酸。
发明有益效果:
本发明提供了一种结构新型的手性吡啶氮氧催化剂,结构丰富,可调节性强。该催化剂原料易得,合成简便、廉价、高效。催化活性高,在O-酰基二氢唑酮不对称Steglich重排反应中,得到α-季碳手性中心C-酰基二氢吡唑酮,取得了高收率和优异的对映选择性产物立体选择性高,收率和对映选择性最高均可达96%,产物经过衍生后可得到具有生理活性的二肽类化合物。
具体实施方式
实施例1
手性4-硝基-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶1-氧化物的合成
在100mL烧瓶中加入L-2-(2,6二异丙基苯酰胺基)吡咯烷(5.48g,20mmol),三乙胺(27.7mL,200mmol,10eq),3-溴4-硝基吡啶氮氧化物(5.45g,25mmol,1.25eq)和四氢呋喃(35mL),60℃搅拌过夜,得到粗产品通过柱层析分离得到亮黄的固体7.83g,产率为95%,>99%ee。CHIRALCEL IA,n-hexane/2-propanol=80/20,flow rate=1.0mL/min,λ=250nm,retention time:10.843min。
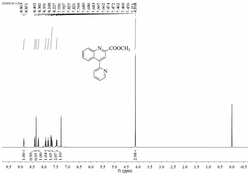
1H NMR(400MHz,CDCl3)δ8.21(s,1H),7.79–7.69(m,2H),7.62(s,1H),7.29(d,J=7.6Hz,1H),7.13(d,J=8.0Hz,2H),4.51(t,J=7.6Hz,1H),3.84(td,J=10.4,5.6Hz,1H),3.01(t,J=8.8Hz,1H),2.91–2.65(m,3H),2.28–2.14(m,2H),2.08–1.94(m,1H),1.17(d,J=6.8Hz,6H),1.01(s,6H).13C NMR(100MHz,CDCl3)δ170.1,146.1,140.6,133.6,130.8,130.0,129.8,128.9,123.6,122.7,64.2,54.0,32.0,29.1,25.9,23.4.
手性4-硝基-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶1-氧化物合成结果
a除非特别说明,反应的步骤如下:3-溴4-硝基吡啶氮氧化物(1.25eq),碱(10eq),b分离收率。
实施例2
手性4-氯-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶1-氧化物的合成
在25mL封管中加入手性4-硝基-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶1-氧化物(2.06g,5mmol),乙酰氯(3.57mL,50mmol,10eq),冰乙酸(10mL)后,升温90-110℃反应,TLC检测反应结束后,将反应液倒入冰水中,用氢氧化钠溶液调制弱碱性,得到的粗产品通过柱层析分离得到棕色固体1.4g,产率为70%,>99%ee。CHIRALCEL IA,n-hexane/2-propanol=90/10,flow rate=1.0mL/min,λ=250nm,retention time:12.632min(minor),27.205min(major)。
1H NMR(600MHz,CDCl3)δ8.09(d,J=2.4Hz,1H),7.84–7.69(m,2H),7.26(d,J=7.2Hz,1H),7.21–7.16(m,1H),7.12(d,J=7.8Hz,2H),4.64–4.58(m,1H),4.11–4.03(m,1H),3.30–3.23(m,1H),2.81(s,2H),2.61–2.52(m,1H),2.29–2.21(m,1H),2.20–2.12(m,1H),2.10–2.01(m,1H),1.14(d,J=6.6Hz,6H),1.04(s,6H).13C NMR(150MHz,CDCl3)δ171.1,146.2,144.5,132.6,131.4,130.5,128.6,127.3,123.5,122.8,64.4,53.7,31.8,29.0,25.1,23.5.
手性4-氯-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶1-氧化物合成结果
a除非特别说明,反应的步骤如下:手性4-硝基-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶1-氧化物(2.06g),冰乙酸(10.0ml),乙酰氯(10eq),b分离收率。
实施例3
手性4-吡咯烷基-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶1-氧化物的合成
在15mL封管中加入手性4-氯-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶氮氧(200mg,0.5mmol)与吡咯烷(1mL)在140℃下反应,反应结束,减压蒸馏后得到粗产品通过柱层析分离得到浅棕色固体133.0mg,产率为61%,>99%ee。CHIRALCEL IA,n-hexane/2-propanol=90/10,flow rate=0.5mL/min,λ=250nm,retention time:20.270min(minor),35.368min(major)。
1H NMR(400MHz,CDCl3)δ8.36(s,1H),8.32(s,1H),7.77(dd,J=7.2,2.0Hz,1H),7.22(t,J=7.6Hz,1H),7.08(d,J=7.6Hz,2H),6.57(d,J=7.2Hz,1H),4.30(t,J=7.2Hz,1H),3.67–3.57(m,1H),3.45–3.34(m,2H),3.25–3.14(m,2H),2.84–2.71(m,3H),2.50–2.41(m,1H),2.28–2.18(m,1H),2.14–2.04(m,1H),2.03–1.93(m,3H),1.91–1.82(m,2H),1.01(d,J=6.8Hz,12H).13C NMR(150MHz,CDCl3)δ171.7,146.2,144.8,135.9,134.1,133.8,131.3,128.2,123.3,111.1,64.0,54.2,49.9,30.4,28.8,25.0,24.2,23.5.
手性4-吡咯烷基-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶氮氧合成结果
a除非特别说明,反应的步骤如下:手性4-氯-3-(2-(2,6二异丙基苯酰胺基)吡咯烷基)吡啶1-氧化物(200mg),吡咯烷(1.0ml),b分离收率。
实施例4
手性4-吡咯烷基-3-(2-(苯酰胺基)吡咯烷基)吡啶氮氧的合成
在15mL的封管中加入手性4-氯-3-(2-(苯酰胺基)吡咯烷基)吡啶氮氧(0.5mmol)与吡咯烷(1mL)在140℃下反应,反应结束,减压蒸馏后得到粗产品通过柱层析分离得到浅棕色固体。
1H NMR(600MHz,CDCl3)δ10.20(d,J=14.4Hz,1H),8.57(s,1H),7.75(d,J=6.6Hz,1H),7.43(t,J=7.2Hz,2H),7.15(td,J=7.8,3.6Hz,2H),6.96(t,J=7.2Hz,1H),6.53(d,J=7.2Hz,1H),4.35(t,J=7.2Hz,1H),3.59–3.51(m,1H),3.48–3.40(m,2H),3.40–3.31(m,2H),2.67–2.60(m,1H),2.41–2.32(m,1H),2.23–2.15(m,1H),2.11–2.00(m,3H),1.98–1.87(m,3H).13C NMR(100MHz,CD3OD)δ172.7,147.8,139.4,137.2,134.2,132.6,129.8,125.3,121.1,112.4,64.7,53.4,51.1,31.4,25.8,24.7.
实施例5
手性4-吡咯烷基-3-(2-(2,6二乙基苯酰胺基)吡咯烷基)吡啶氮氧的合成
在15mL封管中加入手性4-氯-3-(2-(2,6二乙基苯酰胺基)吡咯烷基)吡啶氮氧(0.5mmol)与吡咯烷(1mL)在140℃下反应,反应结束,减压蒸馏后得到粗产品通过柱层析分离得到浅棕色固体。
1H NMR(600MHz,CDCl3)δ9.06(s,1H),8.50–8.46(m,1H),7.67(dd,J=7.2,1.8Hz,1H),7.11(t,J=7.2Hz,1H),6.99(d,J=7.8Hz,2H),6.48(d,J=7.2Hz,1H),4.34(t,J=7.8Hz,1H),3.61–3.54(m,1H),3.44–3.35(m,2H),3.26–3.20(m,2H),2.70(dd,J=16.8,7.8Hz,1H),2.47–2.27(m,5H),2.26–2.17(m,1H),2.13–2.04(m,1H),1.98–1.90(m,3H),1.88–1.79(m,2H),0.95(t,J=7.2Hz,6H).13C NMR(100MHz,CDCl3)δ171.4,144.6,141.6,135.9,133.8,133.5,133.0,127.7,126.3,110.9,63.5,53.8,49.8,30.5,25.0,24.9,24.1,14.5.
实施例6
手性4-吡咯烷基-3-(2-(3,5二三氟甲基苯酰胺基)吡咯烷基)吡啶氮氧的合成
在15mL封管中加入手性4-氯-3-(2-(3,5二三氟甲基苯酰胺基)吡咯烷基)吡啶1-氧化物(0.5mmol)与吡咯烷(1mL)在140℃下反应,反应结束,减压蒸馏后得到粗产品通过柱层析分离得到浅棕色固体。
1H NMR(400MHz,CDCl3)δ8.72(s,1H),8.07(d,J=2.0Hz,1H),7.89(dd,J=7.2,2.0Hz,1H),7.52(s,2H),7.44(s,1H),7.06(d,J=7.2Hz,1H),4.01(q,J=4.4Hz,1H),3.55–3.48(m,1H),3.43–3.70(m,2H),3.33–3.22(m,2H),2.44–2.34(m,1H),2.13–2.02(m,2H),1.99–1.89(m,2H),1.85–1.75(m,4H).13C NMR(150MHz,CDCl3)δ164.9,143.2,139.9,135.2,133.6(d,JC-F=34.5Hz),132.6,123.9,122.8(d,JC-F=271.5Hz),119.3(d,JC-F=3.0Hz),118.0,103.0,97.8,53.0,48.7,46.1,34.4,29.8,27.1,23.8,23.2.
实施例7
手性4-吡咯烷基-3-(2-(苯甲酰胺基)吡咯烷基)吡啶氮氧的合成
在15mL封管中加入手性4-氯-3-(2-(苯甲酰胺基)吡咯烷基)吡啶氮氧(0.5mmol)与吡咯烷(1mL)在140℃下反应,反应结束,减压蒸馏后得到的粗产品通过柱层析分离得到浅棕色固体。
1H NMR(400MHz,CDCl3)δ7.78–7.71(m,2H),7.44(t,J=7.2Hz,2H),7.36(t,J=7.2Hz,1H),7.16–7.10(m,2H),6.47(d,J=6.8Hz,1H),4.05(dd,J=7.6,5.2Hz,1H),3.73–3.65(m,1H),3.31–3.20(m,2H),3.12(s,3H),3.10–3.02(m,2H),2.74–2.66(m,1H),2.08–1.93(m,3H),1.86–1.65(m,5H).13C NMR(150MHz,CD3OD)δ174.3,147.7,144.3,135.8,134.2,133.2,131.1,129.4,128.4,111.7,59.2,51.9,50.5,37.8,31.2,25.7,24.2.
实施例8
手性3,4-二氨基吡啶氮氧催化剂和手性3,4-二氨基吡啶催化剂对O-苄氧羰基-2-(4-甲氧基苯基)-4-苄基二氢唑酮的不对称Steglich重排
在10mL真空管中,加入催化剂(0.0025mmol,5mol%)和 MS(20mg),加入甲苯(0.3mL),将反应液冷却至-40℃,将O-苄氧羰基-2-(4-甲氧基苯基)-4-苄基二氢唑酮(21.0mg,0.05mmol)溶于甲苯(0.2mL)中,加入到反应液中,反应混合物在-40℃条件下反应16h后用0.1M HCl(10mL)淬灭,然后用乙醚(5mL×3)萃取,收集有机相后用Na2SO4干燥,将粗产物用柱层析分离得到产物2-(4-甲氧基苯基)-4-苄基-4-苄氧羰基二氢唑酮产物ee值通过手性HPLC获得。
O-苄氧羰基-2-(4-甲氧基苯基)-4-苄基二氢唑酮的不对称Steglich重排的实验结果
a除非特别说明,反应的步骤如下:催化剂(5mol%),底物浓度(0.05mmol),溶剂体积(0.5mL),b分离收率cee值通过高效液相色谱分离, MS(10mg)。
实施例9
O-苄氧羰基-2-(4-甲氧基苯基)-4-苄基二氢唑酮的不对称Steglich重排
在10mL真空管中,加入催化剂(0.005mmol,5mol%)和 MS(20mg),加入甲苯(0.6mL),将反应液冷却至-40℃,将O-苄氧羰基-2-(4-甲氧基苯基)-4-苄基二氢唑酮(41.5mg,0.1mmol)溶于甲苯(0.4mL)中,加入到反应液中,反应混合物在-40℃条件下反应16h后用0.1M HCl(10mL)淬灭,然后用乙醚(5mL×3)萃取,收集有机相后用Na2SO4干燥,将粗产物用柱层析分离得到产物2-(4-甲氧基苯基)-4-苄基-4-苄氧羰基二氢唑酮产物ee值通过手性HPLC获得。采用如下催化剂对反应进行优化,结果如下:
a除非特别说明,反应的步骤如下:催化剂(5mol%),底物浓度(0.05mmol),溶剂体积(0.5mL),b分离收率cee值通过高效液相色谱分离, MS(10mg)。
实施例10
O-苄氧羰基-2-(4-甲氧基苯基)-4-烷基二氢唑酮的不对称Steglich重排
在10mL真空管中,加入催化剂(2.2mg,0.05mmol,5mol%)和 MS(20mg),加入甲苯(0.6mL),将反应液冷却至-40℃,将O-苄氧羰基-2-(4-甲氧基苯基)-4-烷基二氢唑酮(0.1mmol)溶于甲苯(0.4mL)中,加入到反应液中,反应混合物在-40℃条件下反应16h后用0.1M HCl(10mL)淬灭,然后用乙醚(5mL×3)萃取,收集有机相后用Na2SO4干燥,将粗产物用柱层析分离得到产物2-(4-甲氧基苯基)-4-烷基-4-苄氧羰基二氢唑酮产物ee值通过手性HPLC获得。
O-苄氧羰基-2-(4-甲氧基苯基)-4-烷基二氢唑酮的不对称Steglich重排的实验结果如下:
a除非特别说明,反应的步骤如下:催化剂(5mol%),底物浓度(0.1mmol),溶剂体积(1.0mL), MS(20mg),b分离收率,cee值通过高效液相色谱分离,dN2,20℃。
需要特别指出的是:对于R4为大位阻叔丁基底物(编号11),公开文献中催化体系并没有相关报道。而采用本发明催化体系,也表现出良好的催化性能,重排后也取得了高达90%ee,收率达到66%。
实施例11
在10mL真空管中,加入催化剂(2.2mg,0.05mmol,5mol%)和 MS(20mg),置换氮气,加入甲苯(0.6mL),将反应液冷却至-40℃,将O-苄氧羰基-2-(4-甲氧基苯基)-4-叔丁基二氢唑酮(0.1mmol,38.1g)溶于甲苯(0.4mL)中,加入到反应液中,反应混合物在20℃条件下反应16h后用0.1M HCl(10mL)淬灭,然后用乙醚(5mL×3)萃取,收集有机相后用Na2SO4干燥,将粗产物用柱层析分离得到产物2-(4-甲氧基苯基)-4-烷基-4-苄氧羰基二氢唑酮产物ee值通过手性HPLC获得。
1H NMR(400MHz,CDCl3)δ7.61–7.52(m,2H),7.37–7.28(m,3H),7.25–7.17(m,2H),6.96–6.89(m,2H),5.18(dd,J=36.0,12.4Hz,2H),3.83(s,3H),1.35(s,9H).
13C NMR(150MHz,CDCl3)δ170.7,165.7,163.2,160.7,134.8,128.7,128.6,128.5,127.9,126.8,114.0,102.5,68.5,55.5,35.0,26.8.
实施例12
2-(4-甲氧基苯基)-4-甲基-4-苄氧羰基二氢唑酮产物衍生
2-(4-甲氧基苯基)-4-甲基-4-苄氧羰基二氢唑酮(0.1mmol)溶解在二氯甲烷中(1mL)中并加入到(L)-丙氨酸甲酯盐酸盐(0.15mmol),NEt3(0.2mmol)的混合物中。将DMAP(0.05mmol)溶于二氯甲烷(0.6mL)中,将得到的澄清无色溶液搅拌24h,然后通过快速色谱法纯化产物。
1H NMR(400MHz,CDC13)δ7.78(d,2H,J=7.2),7.65(s,1H),7.26-7.32(m,5H),6.92(d,2H,J=7.2),6.89(d,1H,J=5.6),5.25(d,1H,J=9.6),5.21(d,1H,J=9.6),4.50(pent,1H,J=5.6),3.85(s,3H),3.70(s,3H),1.93(s,3H),1.40(d,3H,J=5.6).13C NMR(100MHz,CDC13)δ172.4,170.4,167.8,166.1,162.5,135.2,129.1,128.4,128.3,128.1,125.9,113.7,68.1,63.2,55.4,52.5,48.8,22.1,17.9.
与现有技术相比,上述方法采用了一种手性3,4-二氨基吡啶氮氧催化剂及其衍生物的合成和手性C-酰基二氢吡唑酮的制备方法;该催化剂原料廉价易得,克服了PPY氮氧与手性酰胺无法直接相连生成手性3,4二氨基吡啶氮氧催化剂的困境,与手性3,4二氨基吡啶相比,此催化剂催化活性高,能够催化O-酰基二氢唑酮发生不对称Steglich重排反应,以很高的产率和对映选择性得到非常有用的α-季碳手性中心的C-酰基二氢吡唑酮。
以上实施例描述了本发明的基本原理、主要特征及优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
3,4-二氨基吡啶氮氧类手性催化剂及其在Steglich重排中的应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。





![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)








动态评分
0.0