

IPC分类号 : C07F9/6568,B01J31/02,C07D209/48,C07B53/00






专利摘要
本发明公开了一类环状膦骨架的手性单膦催化剂Le‑Phos及其全构型的制备方法和应用,所述单膦催化剂为化合物1或化合物1的对映体、消旋体和非对映异构体,化合物1中,“*”表示手性中心;n=0、1、2或3;所述单膦催化剂的制备是:以化合物化合物和化合物为原料,进行取代反应、加成反应、缩合反应、还原反应制备而得。本发明通过使用两种构型的化合物和不同的金属试剂进行加成反应,最后脱保护,可得到所述单膦催化剂1(RP,S,S,RS)、1(SP,R,S,RS)、1(SP,R,R,SS)和1(RP,S,R,SS)的四种全构型的光学纯。本发明还公开了所述单膦催化剂在催化不对称γ‑addition反应中的应用,具有很高的反应活性和立体选择性,具有广泛的应用价值。
权利要求
1.一类环状膦骨架的手性单膦催化剂Le-Phos,其特征在于,所述单膦催化剂为如下所示的化合物1或化合物1的对映体、消旋体或非对映异构体:
化合物1中,R
其中R
2.根据权利要求1所述的单膦催化剂Le-Phos,其特征在于,所述化合物1中的R
3.一种权利要求1所述手性单膦催化剂Le-Phos全构型的制备方法,其特征在于,该方法包括以下具体步骤:
第一步:化合物6溶解在溶剂中,分别与化合物4(R
其中,R
所述溶剂选自干燥的二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯、氯仿或正己烷;
所述缩合反应的温度为-50~100℃;
所述缩合反应的时间为10分钟~48小时;
所述化合物6、化合物4(R
所述缩合剂选自钛酸四乙酯(Ti(OEt)
第二步:在溶剂中,先将BuLi与TMEDA相互作用,生成络合物;然后化合物2在锂络合物作用下生成中间体化合物3
其中,R
所述溶剂选自干燥的二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯、氯仿或正己烷;
所述络合反应的温度为室温~80℃;
所述锂化取代反应的温度为-78~30℃;
所述络合的时间为10分钟~12小时;
所述锂化的时间为10分钟~12小时;
所述BuLi、TMEDA和化合物2的摩尔比为(1~10)∶(1~10)∶(1~10);
所述BuLi为n-BuLi、s-BuLi或t-BuLi;
第三步:化合物7(R
其中,R
所述溶剂选自干燥的二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯、氯仿或正己烷;
所述加成反应的温度为-78~30℃;
所述加成反应的时间为10分钟~12小时;
所述化合物7和化合物3的摩尔比为(1~10)∶(10~1);
第四步:脱保护得到手性单膦催化剂Le-Phos即化合物1(R
其中,R
所述溶剂选自干燥的二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯、氯仿或正己烷;
所述脱保护试剂选自二乙胺、三乙稀二胺、乙醇胺或氢化铝锂;
所述脱保护反应的温度为0~100℃;
所述脱保护反应的时间为10分钟~12小时;
所述化合物1-BH
4.一种权利要求1所述手性单膦催化剂Le-Phos在催化不对称γ-addition反应中合成γ-氨基酸酯化物的应用。
5.根据权利要求4所述的应用,其特征在于,将所述的手性单膦催化剂Le-Phos与联烯酸酯形成季膦盐两性离子化合物,氮亲核试剂亲核进攻,形成两性离子化合物,然后催化剂离去,完成催化循环,合成所述γ-氨基酸酯化物;具体包括:
在惰性气氛下,将手性单膦催化剂Le-Phos和氮亲核试剂加入到有机溶剂中,在-10~50℃下,加入联烯酸酯,在-10~50℃下搅拌,反应0.5~24小时,进行不对称γ-addition反应,合成所述γ-氨基酸酯化物;其中:
所述手性单膦催化剂Le-Phos、氮亲核试剂和联烯酸酯的摩尔比为(0.01~1):(1~100):(1~100)。
6.根据权利要求5所述的应用,其特征在于,所述惰性气氛为氩气或者氮气气氛;所述有机溶剂选自二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯或氯仿。
说明书
技术领域
本发明属于有机化学技术领域,涉及手性单膦催化剂及其制备方法和应用,具体涉及一类一类环状膦骨架的手性单膦催化剂Le-Phos及其制备方法和应用。
背景技术
手性化合物是指分子量、分子结构相同,但左右排列相反,不能重合,如同我们的双手,互成镜像。手性物质的对映体之间有许多相同的理化性质,例如,熔点,溶解度,进行同类型的反应等;手性物质的对映体之间也存在许多不同的理化性质,例如旋光度,绝对构型,与手性化合物反应得到不同的产物等。更重要的是生理活性等可能不同。20世纪5,60年代,曾经使用过的反应停是一种曾在现代医学史上造成巨大灾难的药物,有强烈致畸作用。最终,经研究证实,反应停组分中两种互为对映体的手性分子中的一种具有致畸作用,而另一种分子是安全的。由此可见,合成光学纯的分子对医疗医药,环境,生物,材料具有重要的意义。不对称催化具有使用化学计量的手性催化剂而可能获得大量手性产物,数十年来一直是研究的热点和前沿。在2001年,诺贝尔化学奖就授予在不对称催化氢化、和不对称催化氧化方面做出突出贡献的Knowles、Noyori和Sharpless,标志着不对称催化研究已获得了令人瞩目的成就。
无金属参与,使用手性膦作为催化剂进行不对称催化反应,已经成为绿色化学的重要研究方向,并且在手性医药、农药、香料以及功能材料合成等领域得到广泛应用。在00年之前,由于手性膦催化剂种类少等原因的限制,叔膦催化的不对称反应发展一直比较缓慢。05年之后,大量的手性膦催化剂被合成出来,不对称膦催化方面已经取得了相当大的进展。因此,膦催化不对称反应现在已成为强而有力的工具,用于构建C-C、C-N、C-O和C-S键以及合成多功能团碳环和杂环。
目前,已有报道过的碳手性和硫手性的单膦配体(催化剂)包括有Ming-Phos(Angew.Chem.Int.Ed.2014,53,4350)、Xiao-Phos(Angew.Chem.Int.Ed.2015,54,6874)、Wei-Phos(Angew.Chem.Int.Ed.2015,54,14853)和Peng-Phos(Angew.Chem.Int.Ed.2016,55,13316)等多种C-中心手性新型单膦配体(催化剂)。基于以上基础,在克服了现有技术中合成含膦中心手性的催化剂时,原料昂贵、合成路线冗长、反应试剂毒性大、对映异构体的合成难度大及产率低等缺陷,又发展了一种新型环状膦骨架的、便于高效合成全构型的具有四个手性中心(含C-中心手性、P-中心手性)单膦催化剂。
发明内容
本发明的目的是提供一类手性单膦催化剂Le-Phos及其全构型的制备方法和应用,可高效、简单及低成本的制备全部立体构型的所述手性单膦催化剂Le-Phos。
本发明提供的一类手性单膦催化剂Le-Phos为四中心手性单膦配体,为如下化合物1或、化合物1的对映体、消旋体或非对映异构体:
其中,R
作为一种优选方案,上述化合物1中的R
作为一种优选方案,上述化合物1中的R
作为进一步优选方案,上述化合物1中的R
作为进一步优选方案,上述化合物1中的R
作为更进一步优选方案,所述手性单膦催化剂Le-Phos选自如下化合物或所述化合物的对映体、消旋体或非对映异构体,如下所示:
其中:Ar
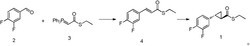
本发明还提供了化合物1全构型的制备方法:
第一步:化合物6溶解在溶剂中,一定温度下分别与化合物4(Rs)、4(Ss)在缩合剂的作用下进行缩合反应,得到化合物7(Rs)、7(Ss),反应过程如下反应式(I)所示:
其中,式(I)中的各基团的定义与化合物1中各基团定义相同。
所述溶剂选自干燥的二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯、氯仿或正己烷;优选地,为干燥的四氢呋喃。
所述缩合反应的温度为-50~100℃;优选地,为50~70℃。
所述缩合反应的时间为10分钟~48小时;优选地,为8小时。
所述化合物6、化合物4和缩合剂的摩尔比为(1~10)∶(1~10)∶(1~10);优选地,为1:1:2。
所述缩合剂的作用为促进缩合反应的进行,选自钛酸四乙酯钛酸四乙酯(Ti(OEt)4)或钛酸四异丙酯、钛酸四甲酯;优选地,为钛酸四异丙酯。
第二步:在溶剂中,先将BuLi与TMEDA相互作用进行络合,生成中络合物;然后化合物2在锂络合物作用下锂化生成中间体化合物3;反应过程如反应式(II)所示:
上述式(II)中的各基团的定义与化合物1中各基团定义相同;式中的n=0,1,2或3;TMEDA为四甲基乙二胺;
所述溶剂选自干燥的二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯、氯仿或正己烷;优选地,为干燥的乙醚。
所述络合反应的温度为室温~80℃;优选地,为50~80℃。
所述取代反应的温度为-78~30℃;优选地,为-78~-50℃。
所述络合的时间为10分钟~12小时;优选地,为0.5~1小时。
所述锂化的时间为10分钟~12小时;优选地,为4~6小时
所述步骤的BuLi、TMEDA和化合物2的摩尔比为(1~10)∶(1~10)∶(1~10);优选地,为2∶2∶1。
所述BuLi的作用为和P邻位氢进行交换、进行取代反应;所述BuLi包括n-BuLi、s-BuLi、t-BuLi。
第三步:化合物7(Rs)、7(Ss)溶解在溶剂中,与中间体化合物3进行加成反应,得到催化剂Le-Phos的硼烷络合物,反应过程如下反应式(III)所示:
上述式(III I)中的各基团的定义与化合物1中各基团定义相同;
所述溶剂选自干燥的二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯、氯仿或正己烷;优选地,为干燥的四氢呋喃。
所述加成反应的温度为-78~30℃;优选地,为-78~-50℃。
所述加成反应的时间为10分钟~12小时;优选地,为6~8小时。
其中,所述化合物7和化合物3的摩尔比为(1~10):(10~1);优选地,为1.5∶1。第四步:脱保护得到手性单膦催化剂Le-Phos即化合物1(RP,S,S,RS)、1(SP,R,S,RS)、1(SP,R,R,SS)和1(RP,S,R,SS),反应过程如下反应式(IV)所示:
上述式(IV)中各基团的定义与化合物1中各基团定义相同。
所述溶剂选自干燥的二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯、氯仿或正己烷;优选地,为干燥的二氯甲烷。
所述脱保护试剂选自二乙胺、三乙稀二胺、乙醇胺或氢化铝锂;优选地,为二乙胺。
所述脱保护反应的温度为0~100℃;优选地,为50~80℃。
所述脱保护反应的时间为10分钟~12小时;优选地,为2~8小时。
所述化合物1-BH3和脱保护试剂的摩尔比为(1~10):(10~1);优选地,为1:4。
本发明方法中,以化合物6: 和化合物4: 为原料,进行缩合反应、与式: 进行加成反应,再脱保护制备所述化合物1手性单膦催化剂Le-Phos。
本发明通过使用两种构型的化合物7和金属试剂加成,可方便地得到手性单膦催化剂Le-Phos四种全构型即化合物1(RP,S,S,RS)、1(SP,R,S,RS)、1(SP,R,R,SS)和1(RP,S,R,SS)的光学纯化合物。
本发明还提供了所述单膦催化剂Le-Phos在催化不对称γ-addition反应中的应用,所述手性单膦催化剂Le-Phos是具有如化合物1的化合物或所述化合物的对映体、消旋体或非对映异构体。
本发明还提供了所述氮亲核试剂参与的不对称γ-addition反应合成γ-氨基酸类衍生物的应用,将所述的手性单膦催化剂Le-Phos与联烯酸酯形成季膦盐两性离子化合物,氮亲核试剂亲核进攻,形成新的两性离子化合物,然后催化剂离去,完成催化循环,合成所述γ-氨基酸类衍生物。所述手性单膦催化剂Le-Phos为化合物1的化合物或所述化合物1化合物的对映体、消旋体或非对映异构体。
如上所述的手性单膦催化剂Le-Phos用于催化不对称γ-addition反应中的应用中,催化氮亲核试剂的不对称γ-addition反应合成γ-氨基酸类衍生物的方法中:
作为一种优选方案,首先使所述手性单膦催化剂Le-Phos与联烯酸酯形成季膦盐两性离子化合物,氮亲核试剂亲核进攻,形成新的两性离子化合物,然后催化剂离去,完成催化循环,合成所述γ-氨基酸类衍生物。反应过程如下反应式(V)所示:
其中,NuH表示氮亲核试剂。
作为进一步优选方案,所述制备包括如下步骤:在惰性气氛下,将所述手性类单膦催化剂Le-Phos与氮亲核试剂加入到有机溶剂中,在-10~50℃搅拌,再加入联烯酸酯在-10~50℃搅拌,反应0.1~24小时,进行不对称γ-addition反应,合成所述γ-氨基酸类衍生物。
作为更进一步优选方案,所述手性单膦催化剂Le-Phos、氮亲核试剂和联烯酸酯的摩尔比为(0.01~1):(1~100):(1~100),以(0.5~1):10:20最佳。
作为更进一步优选方案,所述惰性气氛为氩气气氛或者氮气气氛;所述有机溶剂选自二氯甲烷、乙醚、二丁醚、甲基叔丁基醚、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、甲苯、二甲苯、苯、氯苯、氟苯或氯仿。
所述氮亲核试剂可以是结构如化合物8所示:
所述联烯酸酯可以是结构如化合物9所示:
上述化合物8,化合物9中:R
进一步优选地,R
与现有技术相比,本发明具有如下有益效果:
(1)本发明提供了一类新型手性单膦催化剂,首次报道了所述手性单膦催化剂用于催化氮亲核试剂的不对称γ-addition反应,具有很高的反应活性和立体选择性,可使加成产物: 的产率为54%-98%,对映体过量(ee)为85%-95%。
(2)本发明提供的手性单膦配体的制备方法,克服了现有技术中合成含膦手性配体时,原料昂贵、合成路线冗长、反应试剂毒性大、对映异构体的合成难度大、产率低等缺陷,本发明的制备方法多样且路线短、操作简单,收率为42%-75%,适合规模化生产,具有实用价值。
本发明中:
t-BuLi为叔丁基锂;Ti(O
具体实施方式
结合以下具体实施例,对本发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
下述实施例提供了上述手性单膦催化剂Le-Phos即化合物1的合成方案,具体为:
实施例1 的合成
第一步:在一个100mL的干燥的三口瓶中加入苯甲醛(21.2g,20mmol),叔丁基亚磺酰胺(2.42g,20mmol)和30mL干燥的四氢呋喃,再加入钛酸四异丙酯(5.68g,40mmol),置于80℃中加热回流6h,加水淬灭,过滤,分液,水层用乙酸乙酯萃取三次,合并有机相,分别用水、饱和氯化钠洗涤,无水硫酸钠干燥,过滤,旋干,柱层析纯化,得 (3.98g,95%yield)
第二步:先在一个100mL的干燥的单支口瓶,在氮气氛围下加入干燥的TMEDA和10mL干燥的乙醚,在-50℃下滴加t-BuLi(10mmol,1.3M),搅拌1h;然后向上一操作制备的锂络合物中滴加 (0.89g,5mmol,溶于5mL干燥的乙醚),在-50℃下搅拌4h
其中,t-BuLi为叔丁基锂;TMEDA为四甲基乙二胺。
第三步:向第二步制备的锂化物 溶液中滴加 (1.57g,7.5mmol,溶于10mL干燥的四氢呋喃),在-50℃下搅拌8h,加氯化铵饱和溶液淬灭,分液,水层用乙酸乙酯萃取三次,合并有机相,分别用水、饱和氯化钠洗涤,无水硫酸钠干燥,过滤,旋干,柱层析纯化,得催化剂Le-Phos的硼烷络合物。
第四步:将第三步制备的Le-Phos的硼烷络合物加入到25mL的反应管中,氮气保护,加入4mL二乙胺。在50℃下,搅拌4小时后,降温,旋干,柱层析纯化,得 总产率为72%,比例1.3∶1。
白色固体;[α]
白色固体;[α]
实施例2 b-1(RP,S,S,RS)的个成
具体操作与实施例1相同,仅将所用苯甲醛改用五氟苯甲醛,产率为18%。白色固体;[α]
实施例3 b-1(SP,R,S,RS)的合成
具体操作与实施例1相同,仅将所用苯甲醛改用五氟苯甲醛,产率为24%。白色固体;[α]
实施例4 c-1(RP,S,S,RS)的合成
具体操作与实施例1相同,仅将所用苯甲醛改用对甲基苯甲醛,产率为30%。无色油状物;[α]
实施例5 c-1(SP,R,S,RS)的合成
具体操作与实施例1相同,仅将所用苯甲醛改用对甲基苯甲醛,产率为37%。白色固体;[α]
实施例6 d-1(RP,S,S,RS)的合成
具体操作与实施例1相同,仅将所用苯甲醛改用2-(3,5-二三氟甲基)苯甲醛,总产率为28%。白色固体;[α]
一类环状膦骨架的手性单膦催化剂Le-Phos及其全构型的制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)








动态评分
0.0