专利摘要
本发明属于医药学技术领域,公开了一种抗肿瘤季铵盐类衍生物及其制备方法及应用,经相应的化学反应制备了一系列抗肿瘤衍生物,以及衍生物在抗肿瘤方面的应用。本发明经药理实验表明:所有合成的薯蓣皂苷元季铵盐类衍生物对A549肺癌细胞、H1975肺腺癌细胞、Aspc‑1转移胰腺癌细胞、A431皮肤鳞癌细胞具有明显的抑制作用,抗肿瘤活性均优于薯蓣皂苷元,大部分衍生物对RamosB淋巴瘤细胞抗肿瘤活性优于薯蓣皂苷元。
权利要求
1.一种抗肿瘤季铵盐类衍生物,其特征在于,所述衍生物的分子结构为:
其中X = F、Cl、Br、I。
2.如权利要求1所述的一种抗肿瘤季铵盐类衍生物的制备方法,其特征在于,所述制备方法包括:
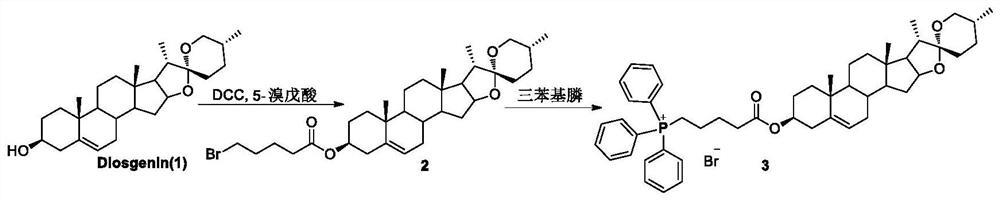
步骤一:将薯蓣皂苷元溶于二氯甲烷,依次加入缩合剂,有机碱,5-溴代戊酸,TLC检测反应完全;减压浓缩后用醇重结晶得化合物3;
所述缩合剂是1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐或者二环己基碳二亚胺;所述有机碱是三乙胺或者吡啶;所述醇是甲醇或者乙醇或者丙醇;
步骤二:所述步骤二为以下方法中任意一种:
将化合物3 溶于乙腈,加入叔胺,80 °C下搅拌反应至完全;将反应液减压浓缩,经硅胶柱色谱分离得到季铵盐类衍生物1a、1d 、1e、 1f、 1h、 1i、 1j、1k;
或者将化合物3溶于乙腈,加入叔胺,80 °C下反应至TLC检测反应完全后,再将反应液减压浓缩,加入正己烷,25 °C下搅拌3 h,抽滤,滤饼为季铵盐类衍生物1b、1c;
或者将化合物3溶于乙腈,加入叔胺,80 °C下反应至TLC检测反应完全后,冷却至室温,析出大量固体,抽滤,滤饼为季铵盐类衍生物1g;
所述薯蓣皂苷元季铵盐类衍生物衍生物的制备方法的反应式为:
。
3.一种如权利要求1所述的抗肿瘤季铵盐类衍生物在制备抗肿瘤药物中的应用,其特征在于,所述肿瘤为与A549肺癌细胞、A431皮肤鳞癌细胞、H1975肺腺癌细胞、Aspc-1转移胰腺癌细胞、Ramos B淋巴瘤细胞相关的肿瘤。
4.一种如权利要求1所述的抗肿瘤季铵盐类衍生物在制备抑制癌细胞药物中的应用,其特征在于,所述癌细胞为A549肺癌细胞、A431皮肤鳞癌细胞、H1975肺腺癌细胞、Aspc-1转移胰腺癌细胞、Ramos B淋巴瘤细胞。
说明书
技术领域
本发明属于医药学技术领域,尤其涉及一种抗肿瘤季铵盐类衍生物及其制备方法及应用。
背景技术
目前,业内常用的现有技术是这样的:
癌症是威胁人类生命和健康的重大疾病,已成为人类死亡的重要因素之一。随着环境与生活方式的变化,我国癌症发病率、死亡率持续上升。因此,探索新型的抗肿瘤药已成为新药研发的重要方向之一。
天然的活性抗肿瘤药物具有疗效显著、毒副作用小等优点。薯蓣皂苷元在自然界中资源丰富,具有抗肿瘤、降血脂、抗血栓、抗炎等多种药理活性。国内外大量研究工作表明,薯蓣皂苷元及其某些衍生物具有明显抗肿瘤作用。以薯蓣皂苷元为先导物进行结构修饰,为寻找更好的抗肿瘤药物提供了一条途径,已受到国内外科学工作者的重视,是一项颇具价值和前景的研究工作。
近年来,科研工作者对薯蓣皂苷元的结构修饰和改造主要集中在 A环与F环上,部分已报道薯蓣皂苷元衍生物具有显著地抗肿瘤活性,但并未完全解决薯蓣皂苷元存在的缺陷,还需从中探索出更优的药物结构。因此,对薯蓣皂苷元进行合理的结构修饰,设计合成新型衍生物,筛选出更加有效的化学结构,仍然具有一定的挑战性。
综上所述,现有技术存在的问题是:
薯蓣皂苷元及其衍生物作为潜在的抗肿瘤活性物质,但其抗肿瘤活性仍然较低缺陷,这在很大程度上限制了其适用范围。
解决上述技术问题的难度和意义:
本发明以增加其抗肿瘤活性为目的,设计并合成了新型的薯蓣皂苷元衍生物,且所有目标衍生物国内外均未报道。为了合成薯蓣皂苷元衍生物,需要在甾体环上引入酯基。
本发明设计将薯蓣皂苷元C3-位羟基通过酯化反应得到羧酸酯,以此为中间体与各种叔胺反应,得到一系列季铵盐类衍生物。
发明内容
为解决上述现有技术存在的问题,本发明的目的在于提供一种薯蓣皂苷元季铵盐类衍生物及其制备方法及应用。
为达到上述目的,本发明的技术方案为:一种抗肿瘤季铵盐类衍生物,其特征在于,所述衍生物的分子结构为:
其中Y为叔胺基或者取代的叔胺基;
X=F、Cl、Br、I。
优选的,所述的一种抗肿瘤季铵盐类衍生物的制备方法,其特征在于,所述制备方法包括:
步骤一:将薯蓣皂苷元溶于二氯甲烷,依次加入缩合剂,有机碱, 5-卤代戊酸,TLC检测反应完全;减压浓缩后用醇重结晶得化合物3;
步骤二:将化合物3溶于乙腈,加入叔胺,80℃下搅拌反应至完全;将反应液减压浓缩,经硅胶柱色谱分离得到季铵盐类衍生物 1a、1d、1e、1f、1h、1i、1j、1k。
优选的,所述一种抗肿瘤季铵盐类衍生物的制备方法,所述步骤一的缩合剂可以是:1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐或者二环己基碳二亚胺;所述有机碱可以是:三乙胺、吡啶等;所述醇可以是:甲醇、乙醇、丙醇;所述步骤二将化合物3溶于乙腈,加入叔胺,80℃下反应至TLC检测反应完全后,再将反应液减压浓缩,加入正己烷,25℃下搅拌3h,抽滤,滤饼为季铵盐类衍生物1b、 1c。
优选的,所述一种抗肿瘤季铵盐类衍生物的制备方法,所述步骤二将化合物3溶于乙腈,加入叔胺,80℃下反应至TLC检测反应完全后,冷却至室温,析出大量固体,抽滤,滤饼为季铵盐类衍生物 1g。
优选的,所述的一种抗肿瘤季铵盐类衍生物的制备方法,其特征在于,所述薯蓣皂苷元季铵盐类衍生物衍生物的制备方法的反应式为:
优选的,所述的抗肿瘤季铵盐类衍生物在制备抗肿瘤药物中的应用。
优选的,所述的抗肿瘤季铵盐类衍生物在制备抑制癌细胞药物中的应用。
优选的,所述的抗肿瘤季铵盐类衍生物在制备抑制癌细胞药物中的应用,其特征在于,所述抑制癌细胞为A549肺癌细胞、A431皮肤鳞癌细胞、H1975肺腺癌细胞、spc-1转移胰腺癌细胞、Ramos B淋巴瘤细胞。
优选的,所述的一种抗肿瘤季铵盐类衍生物,所述抗肿瘤季铵盐类衍生物抗肿瘤活性高于先导物薯蓣皂苷元。
相对于现有技术,本发明的有益效果为:本发明以提高抗肿瘤活性为目的,设计了薯蓣皂苷元季铵盐类衍生物,筛选出了具有较高抗肿瘤活性的甾体化合物。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
现有技术中,在抗肿瘤活性效果差。
本发明实施例提供的薯蓣皂苷元季铵盐类衍生物用下列通式(1) 表示:
其中R1、R2、R3为烃基或取代烃基;
X=F、Cl、Br、I;
本发明实施例提供的薯蓣皂苷元季铵盐类衍生物的制备方法,化学反应式为:
下面结合具体分析对本发明作进一步描述。
1)本发明所有合成的新化合物结构都经1H NMR,13C NMR,IR, HRMS(ESI)确证,用红外、熔点、旋光度表征。所合成的季铵盐类衍生物对A549(人肺癌细胞)、A431(人皮肤鳞癌细胞)、H1975(人肺腺癌细胞)、Aspc-1(人转移胰腺癌细胞)、Ramos(人B淋巴瘤细胞)、进行了相应的生物活性实验。
2)实验所用试剂除特别说明外,都为分析纯试剂,且未经纯化直接使用。1H NMR和13C NMR用Agilent DD2 400-MR(400MHz)核磁共振波谱仪测定,TMS作为内标,CDCl3作为溶剂;IR用FTS 3000傅里叶变换红外光谱仪(美国Digilab公司)测定,KBr压片;HRMS用LCQAdvantage Max质谱仪测定;旋光用SGW-1(上海仪电物理光学仪器有限公司)旋光仪测定;熔点用SGW X-4显微熔点测试仪(上海精密科学仪器有限公司),温度未经校准。
3)细胞培养:
收集A549、A431、H1975、HCT-116、Aspc-1、Ramos的对数生长期细胞,调整细胞悬液浓度,以每孔7×103个细胞,每孔体积100μL 接种到96孔板,每孔设4个复孔(边缘孔用无菌PBS填充)。细胞贴壁后,0%FBS RPMI-1640饥饿8h,对照组用10%FBS RPMI-1640 培养。37℃,5%CO2培养箱中继续培养48h。
4)MTT检测:
在八组细胞分别于培养48h后,加入100μL MTT溶液 (5mg·mL-1),4h后终止培养,每孔加入100μL三联液,于摇床上低速振荡10min,使结晶充分溶解。在酶联免疫检测仪上测定各孔光度值(OD值),选择570nm波长,以无细胞的即RPMl-1640 培养液空白孔调零,测各孔的吸光度值。实验重复三次,记录结果:细胞生长抑制率=(对照组吸光度值-实验组吸光度值)/对照组吸光度值×100%。在GraphPad Prism作图软件中针对抑制剂浓度做图,以便由log[抑制剂]相对于反应,可变斜率模型估算出IC50值。
下面结合具体实施例对本发明作进一步描述。
本发明实施例提供的薯蓣皂苷元季铵盐类衍生物的制备方法,通式(1)化合物的制备路线:
化合物3的合成
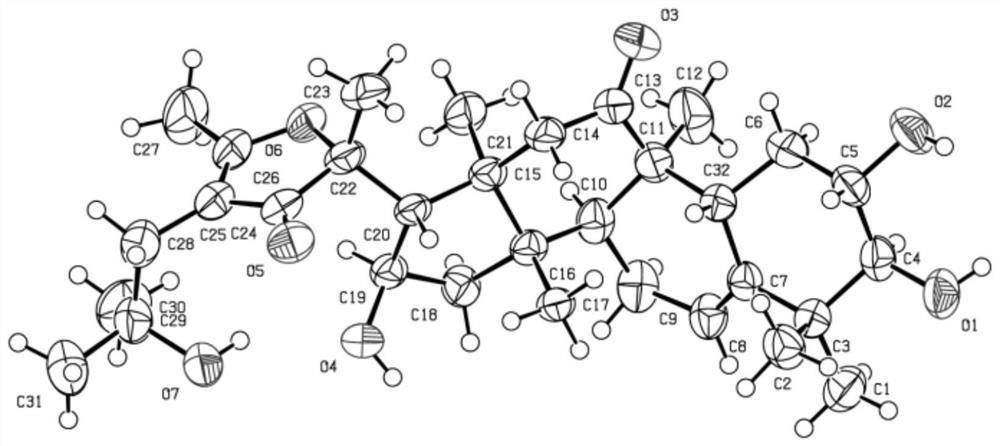
将薯蓣皂苷元(10.0g,24.1mmol)溶于二氯甲烷(300mL),依次加入EDC·HCl(18.4g,96.3mmol),4-二甲氨基吡啶(1.20g,9.82mmol),5-溴戊酸(17.5 g,96.7mmol),25℃反应3h,TLC检测反应完全。反应液依次用2N盐酸(3×100 mL)、饱和碳酸氢钠(3×100mL)、水(3×100mL)洗涤,有机层用无水硫酸钠干燥,减压浓缩后用乙醇重结晶得白色固体3(11.0g,79%)。m.p.108-109℃.[α]18 D–94.3(c 0.003,CHCl3).IR(KBr)νmax 3464,2959,1745,1459,1388,1281,1200, 1052,1010,989,903cm-1.1H NMR(400MHz,CDCl3)δ5.37(d,J=3.4Hz,1H,H-6), 4.68-4.53(m,1H,H-3),4.40(q,J=6.8Hz,1H,H-16),3.52-3.29(m,4H,H-26and -CH2-Br),2.41-2.23(m,4H,H-4and-COCH2-)ppm.13C NMR(100MHz,CDCl3)δ172.66,139.75,122.53,109.40,80.92,74.04,66.96,62.18,56.54,50.04,41.73,40.38,39.84,38.24,37.06,36.85,33.76,33.22,32.16,32.12,31.96,31.51,30.42,28.93,27.90,23.71,20.94,19.47,17.28,16.42,14.67ppm.HR-ESI-MS m/z calcd forC32H49BrO4Na[M+Na]+601.2692,found 601.2690.
化合物1a的合成
将化合物3(0.60g,1.04mmol,1eq.)溶于乙腈(15mL),加入三烯丙基胺 (3.6mL,20.8mmol,20eq.),80℃下搅拌至TLC检测反应完全。将反应液减压浓缩,加入二氯甲烷(30mL)溶解,用5%HBr(3×15mL)洗涤,有机层用无水硫酸钠干燥,减压回收溶剂。经硅胶柱色谱(二氯甲烷:甲醇(v/v)=100:1)分离得到黄色固体1a(0.26g,35%)。m.p.178-179℃.[α]18D–67.0(c 0.002,CHCl3).IR (KBr)νmax 3421,2963,1738,1464,1382,1264,1184,1057,1015,990,962,899cm-1. 1H NMR(400MHz,CDCl3)δ6.10-5.92(m,3H,-CH=CH2 inallyl moiety),5.84-5.55(m, 6H,-CH=CH2 in allyl moiety),5.28(s,1H,H-6),4.49(d,J=5.3Hz,1H,H-3),4.33(d,J= 7.5Hz,1H,H-16),4.08(d,J=6.1Hz,6H,N+-CH2-inallyl moiety),3.39(d,J=10.0Hz, 1H,H-26α),3.27(d,J=10.2Hz,3H,N+-CH2-and H-26β),2.32(m,2H,-COCH2-),2.22 (d,J=7.6Hz,2H,H-4)ppm.13C NMR(100MHz,CDCl3)δ172.36,139.56,129.36, 124.26,122.43,109.26,80.76,74.17,66.81,62.04,61.66,58.57,56.40,49.91,41.57, 40.23,39.68,38.08,36.90,36.70,33.36,32.01,31.80,31.35,30.25,29.66,28.77, 27.73,21.74,21.64,20.79,19.32,17.13,16.26,14.51ppm.HR-ESI-MS m/z calcd for C41H64O4N[M–Br]+634.4830,found 634.4836.
化合物1b的合成
将化合物3(0.80g,1.39mmol,1eq.)溶于乙腈(20mL),加入N,N-二甲基丁胺(3.9mL,27.8mmol,20eq.),80℃下反应至TLC检测反应完全。将反应液减压浓缩,加入正己烷(3×30mL),25℃下搅拌3h,抽滤,滤饼为白色固体 1b(0.81g,86%)。m.p.226-227℃.[α]18D–83.3(c 0.003,CHCl3).IR(KBr)νmax 3461, 2960,1737,1465,1384,1254,1185,1061,1015,987,903cm-1.1H NMR(400MHz, CDCl3)δ5.33(s,1H,H-6),4.54(s,1H,H-3),4.38(d,J=7.2Hz,1H,H-16),3.70-3.40(m, 6H,N+-CH2-and H-26),3.37(s,6H,N+-CH3),2.36(d,J=6.1Hz,2H,-COCH2-), 2.32-2.04(m,4H)ppm.13C NMR(100MHz,CDCl3)δ172.29,139.60,122.53,109.33, 80.84,74.26,66.89,63.96,63.53,62.12,56.48,51.38,49.98,41.65,40.30,39.76, 38.16,36.97,36.78,33.58,32.09,31.88,31.43,30.33,28.85,27.82,24.70,22.15, 21.65,20.86,19.70,19.39,17.20,16.34,14.58,13.79ppm.HR-ESI-MS m/zcalcd for C38H64O4N[M–Br]+598.4830,found 598.4835.
化合物1c的合成
制备方法同1b,得到白色固体1c,产率90%。m.p.248-249℃.[α]18D–78.0 (c0.003,CHCl3).IR(KBr)νmax 3447,2962,1735,1464,1383,1251,1184,1059,1013, 989,905cm-1.1H NMR(400MHz,CDCl3)δ5.33(s,1H,H-6),4.54(d,J=4.5Hz,1H, H-3),4.44-4.30(m,1H,H-16),3.78(s,4H,N+-CH2-in pyrrolidine moiety),3.72-3.59 (m,2H,N+-CH2-),3.43(d,J=9.5Hz,1H,H-26α),3.34(t,J=10.8Hz,1H,H-26β),3.26 (s,3H,N+-CH3)ppm.13CNMR(100MHz,CDCl3)δ172.44,139.63,122.53,109.34, 80.85,74.25,66.89,64.62,63.74,62.13,56.49,49.99,48.85,41.65,40.31,39.77, 38.16,36.98,36.79,33.62,32.10,31.89,31.44,30.34,28.85,27.82,23.41,21.78, 20.87,19.40,17.20,16.34,14.59ppm.HR-ESI-MS m/z calcd for C37H60O4N[M–Br]+ 582.4517,found 582.4521.
化合物1d的合成
制备方法同1a,经硅胶柱色谱(二氯甲烷:甲醇(v/v)=70:1)分离得到白色固体1d,产率47%。m.p.247-249℃.[α]18D–76.3(c 0.003,CHCl3).IR(KBr)νmax 3459,2963,1736,1463,1384,1250,1184,1059,1013,989,904cm-1.1H NMR(400 MHz,CDCl3)δ5.33(s,1H,H-6),4.55(d,J=7.5Hz,1H,H-3),4.37(q,J=7.2Hz,1H, H-16),3.85-3.51(m,6H,N+-CH2-),3.44(d,J=8.4Hz,1H,H-26α),3.39-3.19(m,4H, H-26βand N+-CH3),2.38(s,2H,-COCH2-),2.27(d,J=7.4Hz,2H,H-4)ppm.13C NMR (100MHz,CDCl3)δ172.40,139.62,122.52,109.33,80.83,74.24,66.88,62.11,61.25, 56.48,49.98,41.64,40.30,39.76,38.18,36.98,36.78,33.73,32.09,31.87,31.42, 30.33,28.84,27.85,21.88,21.54,20.89,20.85,20.36,19.41,17.19,16.34,14.58 ppm.HR-ESI-MS m/z calcd for C38H62O4N[M–Br]+596.4673,found 596.4706.
化合物1e的合成
制备方法同1a,经硅胶柱色谱(二氯甲烷:甲醇(v/v)=50:1)分离得到黄色固体1e,产率67%。m.p.232-234℃.[α]18D–63.0(c 0.002,CHCl3).IR(KBr)νmax 3449,2970,2375,1708,1461,1399,1265,1060,801cm-1.1H NMR(400MHz,CDCl3) δ5.36(s,1H,H-6),4.55(s,1H,H-3),4.40(d,J=7.3Hz,1H,H-16),4.33-3.51(m,14H, CH2 in morpholine,N+-CH2-and-CH2-CH2-OH),3.46(d,J=8.6Hz,1H,H-26α),3.36(t, J=10.8Hz,1H,H-26β)ppm.13C NMR(100MHz,CDCl3)δ172.66,139.76,122.59, 109.36,80.90,74.28,66.95,62.25,61.08,60.64,59.64,56.57,55.87,50.06,41.73, 40.39,39.86,38.30,37.10,36.88,33.99,32.21,31.99,31.53,30.42,28.94,28.00, 22.01,21.69,20.97,19.55,17.28,16.44,14.69ppm.HR-ESI-MS m/z calcd for C38H62O6N[M–Br]+628.4572,found628.4565.
化合物1f的合成
制备方法同1a,经硅胶柱色谱(二氯甲烷:甲醇(v/v)=90:1)分离得到黄色固体1f,产率59%。m.p.141-142℃.[α]18D–68.5(c 0.002,CHCl3).IR(KBr)νmax 3438, 2962,1736,1463,1379,1260,1182,1057,986,903,698cm-1.1H NMR(400MHz, CDCl3)δ7.90(d,J=8.1Hz,2H,H-Ph),7.56(t,J=7.6Hz,2H,H-Ph),7.46(t,J=7.2Hz, 1H,H-Ph),5.26(s,1H,H-6,),4.50-4.28(m,4H,N+-CH2-,H-3and H-16),3.91(s,6H, N+-CH3),3.41(d,J=8.2Hz,1H,H-26α),3.31(t,J=10.9Hz,1H,H-26β),2.20(t,J=6.7 Hz,2H,-COCH2-),2.13(d,J=7.7Hz,2H,H-4)ppm.13C NMR(100MHz,CDCl3)δ 172.12,144.34,139.57,130.82,130.41,122.37,120.85,109.27,80.78,73.97,68.31, 66.82,62.07,56.42,55.21,55.15,49.91,41.59,40.24,39.70,38.01,36.88,36.69, 33.64,32.02,31.82,31.37,30.28,28.79,27.66,22.97,21.37,20.80,19.33,17.15, 16.28,14.54ppm.HR-ESI-MS m/z calcd forC40H60O4N[M–Br]+618.4517,found 618.4501.
化合物1g的合成
将化合物3(0.50g,0.87mmol,1eq.)溶于乙腈(20mL),加入N,N-二甲基苄胺(2.6mL,17.4mmol,20eq.),80℃下搅拌至TLC检测反应完全,冷却至室温,析出大量固体,抽滤,滤饼为白色固体1g(0.55g,77%)。m.p.250-251℃.[α]18D –79.9(c 0.003,CHCl3).IR(KBr)νmax 3459,2964,1730,1463,1388,1257,1178,1057, 1014,986cm-1.1H NMR(400MHz,CDCl3)δ7.64(d,J=6.7Hz,2H,H-Ph),7.49-7.35 (m,3H,H-Ph),5.34(d,J=3.6Hz,1H,H-6),5.02(s,2H,N+-CH2-Ph),4.61-4.49(m,1H, H-3),4.38(q,J=7.4Hz,1H,H-16),3.64-3.52(m,2H,N+-CH2-),3.48-3.41(m,1H, H-26α),3.34(t,J=10.9Hz,1H,H-26β),3.29(s,6H,N+-CH3),2.35(dd,J=14.2,7.2Hz, 3H),2.27(d,J=7.8Hz,2H)ppm.13C NMR(100MHz,CDCl3)δ172.30,139.61,133.28, 130.81,129.30,127.29,122.52,109.33,80.83,74.23,67.44,66.88,63.18,62.12, 56.48,49.97,49.87,41.65,40.30,39.76,38.15,36.97,36.77,33.57,32.09,31.88, 31.43,30.33,28.84,27.81,22.30,21.66,20.86,19.39,17.20,16.34,14.59ppm. HR-ESI-MS m/z calcd for C41H62O4N[M–Br]+632.4673,found632.4657.
化合物1h的合成
制备方法同1a,经硅胶柱色谱(二氯甲烷:甲醇(v/v)=40:1)分离得到白色固体1h,产率75%。m.p.256-257℃.[α]18D–77.7(c 0.003,CHCl3).IR(KBr)νmax 3459,2963,1735,1464,1403,1268,1189,1062,987cm-1.1H NMR(400MHz,CDCl3) δ5.34(s,1H,H-6),4.63-4.49(m,1H,H-3),4.38(q,J=7.3Hz,1H,H-16),3.69-3.11(m, 10H,N+-CH2-and H-26),2.40(t,J=6.5Hz,2H,-COCH2-),2.28(d,J=8.0Hz,2H,H-4) ppm.13C NMR(100MHz,CDCl3)δ172.26,139.51,122.41,109.22,80.73,74.13, 66.77,62.03,57.09,56.38,53.53,49.88,41.54,40.20,39.65,38.05,36.87,36.67, 33.40,31.98,31.77,31.33,30.22,28.74,27.70,21.67,21.33,20.75,19.28,17.10, 16.23,14.48,8.04ppm.HR-ESI-MS m/z calcd for C38H64O4N[M–Br]+598.4830,found 598.4813.
化合物1i的合成
制备方法同1a,经硅胶柱色谱(二氯甲烷:甲醇(v/v)=50:1)分离得到白色固体1i,产率65%。m.p.219-220℃.[α]18D–85.0(c 0.003,CHCl3).IR(KBr)νmax 3426, 2962,1736,1462,1388,1257,1182,1058,900cm-1.1H NMR(400MHz,CDCl3)δ5.29 (s,1H,H-6),4.52(s,1H,H-3),4.37(s,2H,H-16and-CH-OH),3.98(s,2H,N+-CH2-), 3.72-3.14(m,8H,N+-CH2-and H-26)ppm.13C NMR(100MHz,CDCl3)δ172.59, 139.70,122.52,109.35,80.87,74.22,66.92,64.14,63.89,62.17,56.52,55.08,54.21, 50.00,41.68,40.34,39.79,38.18,37.02,36.82,33.78,32.14,31.92,31.47,30.37, 28.89,27.85,26.41,21.87,21.83,21.68,20.91,19.45,18.19,17.24,16.39,14.64ppm. HR-ESI-MS m/z calcd forC39H62O5N[M–Br]+624.4623,found 624.4609.
化合物1j的合成
制备方法同1a,经硅胶柱色谱(二氯甲烷:甲醇(v/v)=70:1)分离得到黄色固体1j,产率63%。m.p.244-245℃.[α]18D–78.7(c 0.003,CHCl3).IR(KBr)νmax 3452, 2952,2378,1732,1521,1459,1405,1179,1054cm-1.1H NMR(400MHz,CDCl3)δ 5.36(s,1H,H-6),4.58(s,1H,H-3),4.40(d,J=7.2Hz,1H,H-16),4.11(s,2H,-CH2-OH), 3.90-3.00(m,10H,N+-CH2-and H-26)ppm.13C NMR(100MHz,CDCl3)δ172.46, 139.70,122.57,109.39,80.90,74.30,66.95,62.19,61.10,60.37,58.94,56.55,55.49, 50.05,41.72,40.37,39.83,38.22,37.04,36.85,33.62,32.16,31.95,31.50,30.40, 28.91,27.87,21.82,21.35,21.05,20.93,20.06,19.46,17.25,16.40,14.64ppm. HR-ESI-MS m/z calcd for C39H64O5N[M–Br]+626.4779,found 626.4779.
化合物1k的合成
制备方法同1a,经硅胶柱色谱(二氯甲烷:甲醇(v/v)=50:1)分离得到黄色固体1k,产率73%。m.p.222-223℃.[α]18D–62.0(c 0.003,CHCl3).IR(KBr)νmax 3443,2958,2378,1732,1461,1185,1057,902cm-1.1H NMR(400MHz,CDCl3)δ5.36 (s,1H,H-6),4.55(s,1H,H-3),4.39(q,J=7.4Hz,1H,H-16),4.28-3.50(m,13H,CH2 in morpholine,N+-CH2-andN+-CH3),3.46(d,J=7.6Hz,1H,H-26α),3.35(t,J=10.9Hz, 1H,H-26β)ppm.13C NMR(100MHz,CDCl3)δ172.43,139.56,122.47,109.24,80.75, 74.20,66.80,65.22,62.06,61.15,60.26,56.41,49.90,48.50,41.57,40.22,39.69, 38.15,36.93,36.72,33.81,32.03,31.81,31.36,30.25,28.77,27.85,21.86,21.58, 20.79,19.36,17.11,16.26,14.51ppm.HR-ESI-MS m/z calcd for C37H60O5N[M–Br]+ 598.4466,found 598.4471.
薯蓣皂苷元季铵盐衍生物体外抗肿瘤活性测试结果如下表所示:
从抗肿瘤活性测试数据中可以反映:所合成的化合物1a-1k的抗肿瘤活性比先导物2的活性更强。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何不经过创造性劳动想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书所限定的保护范围为准。
一种抗肿瘤季铵盐类衍生物及其制备方法及应用专利购买费用说明

Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

























动态评分
0.0