








专利摘要
本发明属于化合物合成领域,具体涉及一种二苯乙烯类衍生物的镧系稀土金属配合物及其制备方法和应用。所述二苯乙烯类衍生物的镧系稀土金属配合物的结构如式(Ⅰ)所示,式(I)中,M为Eu3+或La3+。本发明提供的二苯乙烯类衍生物的镧系稀土金属配合物不仅能够保留二苯乙烯类衍生物的可逆E/Z异构化特性,而且还能够促进镧系金属的发光性能,为一种集E/Z异构化与光功能特性于一体的多功能光学材料,其在分子开关材料领域具有良好的应用潜力。
权利要求
1.一种二苯乙烯类衍生物的镧系稀土金属配合物,其特征在于,所述二苯乙烯类衍生物的镧系稀土金属配合物的结构如式(Ⅰ)所示:
式(I)中,M为Eu
2.权利要求1所述的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,其特征在于,该方法包括以下步骤:
(1)将芪-4-甲酸乙酯和水合肼在90~110℃下搅拌加热回流反应5~15h,冷却析出沉淀,并将该沉淀洗涤、干燥,得到中间产物L
(2)将所述中间产物L
(3)将所述中间产物L
3.根据权利要求2所述的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,其特征在于,步骤(1)中,所述芪-4-甲酸乙酯和水合肼的摩尔比为(0.001~0.1):1。
4.根据权利要求2所述的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,其特征在于,步骤(2)中,所述中间产物L
5.根据权利要求2所述的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,其特征在于,步骤(2)中,所述中间产物L
6.根据权利要求2所述的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,其特征在于,步骤(1)中,所述洗涤所采用的溶剂为水;步骤(2)中,所述洗涤所采用的溶剂为水和乙醇的混合溶剂。
7.根据权利要求2~6中任意一项所述的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,其特征在于,步骤(3)中,所述中间产物L
8.根据权利要求2~6中任意一项所述的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,其特征在于,步骤(3)中,所述中间产物L
9.根据权利要求2~6中任意一项所述的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,其特征在于,步骤(3)中,所述纯化的方式为采用甲醇和正己烷的混合溶液进行重结晶,过滤,干燥。
10.权利要求1所述的二苯乙烯类衍生物的镧系稀土金属配合物作为分子开关材料的应用。
说明书
技术领域
本发明属于化合物合成领域,具体涉及一种二苯乙烯类衍生物的镧系稀土金属配合物及其制备方法和应用。
背景技术
二苯乙烯类衍生物具有很好的光学活性,研究表明,二苯乙烯类衍生物与有机小分子的配合物在一定光照条件下会发生可逆的E/Z异构化现象,基于这一光学特性,使其在光电信息储存材料、分子开光、生物化学和超分子化学等领域具有巨大的应用潜力。
例如,Patrícia P等人合成了β二酮和光活性t-bpete(反式-1,2-双(4-吡啶基)乙烯)配位的铕与钆的配合物(Lima,P.P.;Nolasco,M.M.;Paz,F.A.A.;Ferreira,R.A.S.;Longo,R.L.;Malta,O.L.;Carlos,L.D.,Photo-Click Chemistry to Design HighlyEfficient Lanthanide β-Diketonate Complexes Stable under UVIrradiation.Chemistry of Materials 2013,25(4),586-598),研究发现在UV-A曝光(>330nm)期间,两种配合物的发射强度急剧增加20倍,而对于t-Eu,发射量子产率至少增加30倍,并进行B3LYP量化计算,计算结果和实验表明在UV-A曝光下的bpete配体的反式-顺式异构化的光照状态一致。其中,这里涉及的配体为吡啶基乙烯。
又如,Poulami Pal等人合成了二芳基乙烯单核Ru(II)三联吡啶配合物(Pal,P.;Mukherjee,S.;Maity,D.;Baitalik,S.,Synthesis,Structural Characterization,andLuminescence Switching of Diarylethene-Conjugated Ru(II)-TerpyridineComplexes by trans-cis Photoisomerization:Experimental and DFT/TD-DFTInvestigation.Inorg Chem 2018,57(10),5743-5753),研究发现随着二芳基乙烯在UV可见光的光照下发生了反式-顺式光致异构化,其配合物的吸收和发光光谱发生了显著变化,再用可见光或加热处理后,可以实现从顺式到反式的回复。其中,这里涉及的配体为一系列二苯乙烯基对位取代的三联吡啶衍生物。
综上,现有的二苯乙烯类配合物的配体基本均为小分子有机物,而并没有做任何与稀土金属相关的研究。
发明内容
本发明旨在提供一种新的二苯乙烯类衍生物的镧系稀土金属配合物及其制备方法和应用。
具体地,本发明提供了一种二苯乙烯类衍生物的镧系稀土金属配合物,其中,所述二苯乙烯类衍生物的镧系稀土金属配合物的结构如式(Ⅰ)所示:
式(I)中,M为Eu
本发明还提供了所述二苯乙烯类衍生物的镧系稀土金属配合物的制备方法,该方法包括以下步骤:
(1)将芪-4-甲酸乙酯和水合肼在90~110℃下搅拌加热回流反应5~15h,冷却析出沉淀,并将该沉淀洗涤、干燥,得到中间产物L1;
(2)将所述中间产物L1、2-氯甲基吡啶盐酸盐和氢氧化钠溶于溶剂中,之后在70~90℃下搅拌加热回流反应5~15h,冷却析出沉淀,并将该沉淀洗涤、干燥,得到中间产物L2;
(3)将所述中间产物L2和苯甲酰三氟丙酮溶于溶剂中,再将所得溶液的pH值调节至7~8,之后滴加含有M的盐酸盐的溶液,M为Eu
进一步的,步骤(1)中,所述芪-4-甲酸乙酯和水合肼的摩尔比为(0.001~0.1):1。
进一步的,步骤(2)中,所述中间产物L1和2-氯甲基吡啶盐酸盐的摩尔比为1:(1~3)。
进一步的,步骤(2)中,所述中间产物L1和氢氧化钠的摩尔比为1:(3~5)。
进一步的,步骤(1)中,所述洗涤所采用的溶剂为水。
进一步的,步骤(2)中,所述洗涤所采用的溶剂为水和乙醇的混合溶剂。其中,水和乙醇的体积比可以为(1~2):1。
进一步的,步骤(3)中,所述中间产物L2和苯甲酰三氟丙酮的摩尔比为1:(1~3)。
进一步的,步骤(3)中,所述中间产物L2和M的盐酸盐的摩尔比为1:(0.5~2)。
进一步的,步骤(3)中,所述纯化的方式为采用甲醇和正己烷的混合溶液进行重结晶,过滤,干燥。其中,所述混合溶液中甲醇和正己烷的体积比可以为(1~2):1。
进一步的,步骤(2)和步骤(3)中所采用的溶剂可以为现有的各种能够作为反应介质的惰性液态物质,其具体实例包括但不限于:水、醇类溶剂、酯类溶剂、醚类溶剂等中的至少一种。此外,步骤(2)中,将所述中间产物L1、2-氯甲基吡啶盐酸盐和氢氧化钠溶于溶剂中的方式没有特别的限定,例如,可以将中间产物L1和2-氯甲基吡啶盐酸盐先溶于溶剂中,之后再滴加氢氧化钠水溶液。
此外,本发明还提供了所述二苯乙烯类衍生物的镧系稀土金属配合物作为分子开关材料的应用。
本发明首次报导含二苯乙烯类衍生物的镧系金属配合物及其制备和性质研究,发现二苯乙烯类衍生物与镧系金属配位,不仅能够保留二苯乙烯类衍生物的可逆E/Z异构化特性,而且还能够促进镧系金属的发光性能,所得配合物为集E/Z异构化与光功能特性于一体的多功能光学材料,其在分子开关材料领域具有良好的应用潜力。此外,本发明提供的二苯乙烯类衍生物的镧系稀土金属配合物的制备方法简单、反应条件温和、原料易得、易于操作,适于推广应用。
附图说明
图1为实施例1所得中间产物L1的
图2为实施例1所得中间产物L2的
图3为实施例1所得目标产物La(tfd)2HL·CF3COO配合物的
图4为实施例2所得目标产物Eu(tfd)2HL·CF3COO配合物的
图5为测试例1中,La(tfd)2HL·CF3COO配合物在乙腈(MeCN)和乙醇(EtOH)溶液中的光致紫外-可见吸收光谱变化图;
图6a和图6b为测试例2中,La(tfd)2HL·CF3COO配合物在乙腈和乙醇溶液中在紫外灯UV-312nm和UV-254nm的循环照射下的紫外-可见吸收光谱变化图;
图7a和图7b为测试例3中,Eu(tfd)2HL·CF3COO配合物在乙腈和乙醇溶液中以及固体状态下的荧光发射光谱。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例旨在用于解释本发明,而不能理解为对本发明的限制。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1
(1)将芪-4-甲酸乙酯(2mmol,0.50g)加到30mL的水合肼中,在100℃下搅拌加热回流反应10h,冷却析出沉淀,将所得沉淀用水洗涤,干燥,得到中间产物L1;
所述中间产物L1的各项参数如下:
产率:65%,熔程:197~198℃,其
(2)将1mmol的中间产物L1(0.24g)和2mmol的2-氯甲基吡啶盐酸盐(0.328g)溶于适量的水和乙醇按照质量比1:1的混合溶液中,微热使其全部溶解,在80℃下将含有4mmol的氢氧化钠(0.36g)水溶液滴加到上述溶液中,将温度控制在80℃下搅拌10h,冷却后析出沉淀,用水和乙醇按照质量比1:1的混合溶剂洗涤,真空干燥,得到中间产物L2;
所述中间产物L2的各项参数如下:
产率:70%,熔程:166~168℃,其
(3)称取0.84g的中间产物L2(0.2mmol)和0.86g的苯甲酰三氟丙酮(0.4mmol)溶于20mL的四氢呋喃中,微热回流使其全部溶解,加入氢氧化钠的水溶液将pH值调节至7~8,加入含有La(Cl3)3·6H2O(0.2mol,0.71g)的甲醇溶液,将温度控制在65℃下回流加热12h,趁热过滤,将所得溶液旋蒸后,用甲醇和正己烷按照质量比1:1的混合溶液重结晶,过滤,干燥,得到的目标产物La(tfd)2HL·CF3COO配合物,产率:70%。
La(tfd)2HL·CF3COO配合物的
FT-IR(cm
元素分析:LaC49H36F9N4O7,理论值%:N:5.081;C:53.37;H:3.291,实测值%:N:5.15;C:53.38;H:3.50。
实施例2
按照实施例1的方法制备二苯乙烯类衍生物的镧系稀土金属配合物,不同的是,将La(Cl3)3·6H2O采用相同摩尔量的Eu(Cl3)3·6H2O替换,得到目标产物Eu(tfd)2HL·CF3COO配合物。
所述产物Eu(tfd)2HL·CF3COO配合物的各项参数如下:
产率:74%;
Eu(tfd)2HL·CF3COO配合物的
FT-IR(cm
元素分析:Eu C49H36F9N4O7,理论值%:N:5.021;C:52.745;H:3.252,实测值%:N:5.02;C:52.75;H:3.24。
实施例1和实施例2中各物质的结构式如表1所示:
表1
测试例1:测试实施例1所得配合物La(tfd)2HL·CF3COO的光致紫外-可见吸收光谱
(1)溶液的配制:室温用DMSO配制浓度为1.0×10
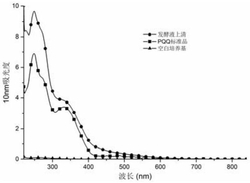
(2)测试方法:测试溶液盛在1cm石英比色皿中,用312nm紫外光照射一定时间后扫描紫外可见吸收光谱,光照到光谱不变后的溶液选择波长254nm紫外光照测试回复的吸收光谱,所得结果如图5所示,其中,a为La(tfd)2HL·CF3COO的乙醇溶液在312nm紫外光照射不同时间后的吸收光谱,b为La(tfd)2HL·CF3COO的乙醇溶液在312nm紫外光照射一定时间后再在254nm紫外光照射一定时间后的吸收光谱,c为La(tfd)2HL·CF3COO的乙腈溶液在312nm紫外光照射不同时间后的吸收光谱,d为La(tfd)2HL·CF3COO的乙腈溶液在312nm紫外光照射一定时间后再在254nm紫外光照射一定时间后的吸收光谱。从图5中a~d的结果可以看出,配合物La(tfd)2HL·CF3COO在乙醇和乙腈溶液中紫外吸收光谱的变化相似,在紫外灯UV-312nm的照射下,位于325nm处的π-π*跃迁的特征吸收峰强度随光照时间的增长逐渐减小,同时位于254nm左右的π-π*跃迁的特征吸收峰强度逐渐增强,并且在270nm处出现了一个等吸收点;配合物La(tfd)2HL·CF3COO在50min后达到光稳态,说明含二苯乙烯基团部分实现了反式到顺式的异构化转变,之后在紫外灯UV-254nm的照射下,π-π*跃迁的特征吸收峰强度逐渐增大,实现了顺式到反式的转变,在240min后达到了一个新的光稳态。
测试例2:测试实施例1所得配合物La(tfd)2HL·CF3COO在紫外灯UV-312nm和UV-254nm的循环照射下的紫外-可见吸收光谱变化图。
(1)溶液的配制:室温用DMSO配制浓度为1.0×10
(2)测试方法:测试溶液盛在1cm石英比色皿中,测量未光照的紫外可见吸收光谱图;然后用312nm紫外光照射10min后扫描紫外可见吸收光谱,再用波长254nm紫外光照照射15min后测试回复的吸收光谱;然后一样条件下得到循环光照下的紫外-可见吸收光谱变化图,所得结果如图6a和图6b所示,其中,图6a为在乙醇溶液中对应的结果,图6b为在乙腈溶液中对应的结果。从图6a和图6b可以看出,在循环光照的过程中,配合物La(tfd)2HL·CF3COO在交替的紫外灯照射下,随着光照时间的增长,发生部分的其他光化学反应,配合物La(tfd)2HL·CF3COO的吸光度逐渐减小,但仍保持着其可循环性,其中在乙腈溶液中的循环性更佳。
测试例3:测试实施例2所得配合物Eu(tfd)2HL·CF3COO的荧光发射光谱
(1)溶液的配制:室温用DMSO配制浓度为1.0×10
(2)测试方法:测试溶液盛在1cm石英比色皿中用最大激发波长得到样品的荧光发射谱图,所得结果如图7a和图7b所示,图7a为在乙醇和乙腈溶液中对应的结果,图7b为Eu(tfd)2HL·CF3COO固体粉末状态下对应的结果。从图7a和图7b可以看出,可观察到配合物的发射光谱均呈现出中心离子Eu(Ⅲ)的特征跃迁。配合物Eu(tfd)2HL·CF3COO在350nm的激发波长下,在579nm、590nm、614nm和651nm左右有四个发射峰,可分别归属Eu(Ⅲ)特有的5D0→7FJ(J=0-3)能级跃迁。其中,位于614nm处的窄带强发射峰,对应于5D0→7F2能级的4f电子跃迁,为超灵敏电偶极跃迁,其荧光发射峰比较尖锐,强度最大。由于配合物Eu(tfd)2HL·CF3COO在溶剂中时具有溶剂效应使荧光猝灭,故在固体状态下的荧光强度相对比乙醇和乙腈溶液中的强。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在不脱离本发明的原理和宗旨的情况下在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
二苯乙烯类衍生物的镧系稀土金属配合物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











![一种基于邻羧基苯乙酸配体的[CdNa]异金属荧光材料及其制备方法](https://www.zhichawang.com/images/ui/CN2019105683801/CN2019105683801.jpg)







动态评分
0.0