









专利摘要
本发明公开了一种高速逆流色谱结合制备液相色谱从钩吻生物碱中分离钩吻生物碱单体的方法,该方法是将钩吻提取物采用高效液相色谱确定目标钩吻生物碱单体组分后,依次通过高速逆流色谱法初步分离和制备液相色谱法二次分离,分别收集各目标钩吻生物碱单体组分。该方法采用多个色谱仪联用分离钩吻单体,具有制备量大,周期短,纯度高等优点,可同时分离获得8个钩吻生物碱单体,具备可控性和自动化生产性,且具有广泛的应用前景。
权利要求
1.一种高速逆流色谱结合制备液相色谱从钩吻生物碱中分离钩吻生物碱单体的方法,其特征在于:将钩吻提取物采用高效液相色谱确定目标钩吻生物碱,依次通过高速逆流色谱法初步分离和制备液相色谱法二次分离,分别收集各目标钩吻生物碱单体组分;
所述高速逆流色谱法的条件:
采用的两相体系由0.5~1.5wt%三乙胺水溶液、正己烷、乙酸乙酯、乙醇按体积比3~5:1~3:2~4:1~3组成;
高速逆流色谱仪参数:转速为800~900rpm/min,柱温箱为20~30℃,流速为1.5~2.5mL/min;
在高速逆流色谱进行初步分离过程中,依据高速逆流色谱图谱收集各目标钩吻生物碱单体组分:流分I:80~100min;流分II:128~143min;流分III:187~230min;流分IV:249~298min;高速逆流色谱图谱采集至400min时,管柱内溶液上下相分离,上相为流分V,下相为流分VI;
所述制备液相色谱法的条件:
色谱柱:UnitaryC18反相色谱柱;
采用的流动相体系由0.4~0.6wt%三乙胺-0.1~0.3wt%冰乙酸混合溶液和乙腈组成;
采用梯度洗脱程序,依据制备液相色谱图谱收集各目标钩吻生物碱单体组分:
流分I:0~50min:乙腈体积百分比由13%匀速升高至50%,依次收集得到羟基钩吻素己和常绿钩吻碱;
流分II:0~25min:乙腈体积百分比由22%匀速升高至30%;25~35min:乙腈体积百分比由30%匀速升高至60%;35~40min:乙腈体积百分比保持在60%,依次收集得到常绿钩吻碱和羟基胡蔓藤碱乙;
流分III:0~10min:乙腈体积百分比由30%匀速升高至40%;10~30min:乙腈体积百分比由40%匀速升高至50%;30~35min:乙腈体积百分比保持在50%,收集得到钩吻素子;
流分IV:0~40min:乙腈体积百分比由25%匀速升高至60%;40~45min:乙腈体积比保持在60%,收集得到钩吻素甲;
流分V:0~25min:乙腈体积百分比由30%匀速升高至45%;25~35min:乙腈体积百分比保持在45%;依次收集得到钩吻绿碱及11-羟基胡蔓藤碱乙;
流分VI:0~40min:乙腈体积百分比由30%匀速升高至50%;40~45min:乙腈体积百分比保持在50%,依次收集得到钩吻绿碱和胡蔓藤碱乙;
制备液相色谱参数:流速为4~8mL/min。
2.根据权利要求1所述的一种高速逆流色谱结合制备液相色谱从钩吻生物碱中分离钩吻生物碱单体的方法,其特征在于:
所述高效液相色谱法的条件:
色谱柱:安捷伦ZORBAX SB-C18色谱柱;
流动相:0.1~0.3wt%磷酸-0.4~0.6wt%三乙胺混合水溶液和乙腈;
采用梯度洗脱程序:0~65min,乙腈体积百分比由12%匀速升高至47%;
高效液相色谱参数:流速为0.8~1.2mL/min;柱温为28~32℃;进样量为15~25μL。
说明书
技术领域
本发明涉及一种从钩吻生物碱中分离钩吻生物碱单体的方法,特别涉及一种联合应用高速逆流色谱法和制备液相色谱法从钩吻生物碱中分离制备钩吻生物碱单体的方法,属于天然药物活性成分分离技术领域。
背景技术
钩吻(Gelsemium elegans)又名断肠草、大茶药、大炮叶、黄花苦蔓、黄猛菜等,为马钱科钩吻属常绿木质藤本植物,产于江西、福建、台湾、湖南、广东、海南、广西、贵州、云南等省区,具有消肿止痛、拔毒杀虫之效,常用作中兽医草药,对猪、牛、羊有驱虫功效,亦可作农药,防治水稻螟虫(中国科学院《中国植物志》编辑委员会,中国植物志,第61卷[M],北京:科学出版社,1992:251- 253.)。钩吻有大毒,为世界著名的有毒植物。但由于钩吻的根茎叶都可入药,研究者们对钩吻的研究在药物代谢和残留、生理和毒理方面越来越深。研究表明,生物碱是钩吻中的主要活性成分,它们具有显著的镇痛、镇静、抑制血小板聚集、促进造血、抗肿瘤和免疫调节等作用(张志阳;伍勇;孙志良,钩吻药效学的研究进展[J],中国兽医杂志,2018,54(04),57-60)。对钩吻生物碱化学成分进行更深入的研究,挖掘新天然产物,有助于提高钩吻植物资源的综合利用,有助于钩吻在植物学、植物化学、药理学、毒理学等方面的研究,同时为新活性成分的寻找提供基础。
目前,已经鉴定出190多种具有复杂结构特征和多种生物效应的化合物,其中包括120多种具有吲哚或羟吲哚核生物碱(A.Ghosh,R.G.Carter,Recent Syntheses andStrategies toward Polycyclic Gelsemium Alkaloids.AngewChemInt Ed Engl58,681-694(2019))。文献对钩吻生物碱的分离多采用传统的硅胶柱层析法。硅胶柱层析法以硅胶为吸附剂,具有不可逆吸附,耗时长,操作繁琐,分离效率低,重现性差的缺点,同时,对一些低含量成分难以获得(Zhou X,Liang J,Zhang Y,et al.Separation and purificationofα-glucosidase inhibitors from Polygonatum odoratum by stepwise high-speedcounter-current chromatography combined with Sephadex LH-20 chromatographytarget-guided by ultrafiltration–HPLC screening[J]. Journal of ChromatographyB,2015,985:149-154)。因此,建立一种高效、重现性好、制备化合物数量多的钩吻生物碱单体的方法,对钩吻进一步临床研究及质量控制具有重要意义。
高速逆流色谱(HSCCC)是近些年来发展起来的一种液-液色谱分离技术,固定相和流动相均为液体,无不可逆吸附,具有高效、快速、重现性好等优点,已广泛应用于天然化合物的分离纯化。一些研究者通过高速逆流色谱法从钩吻中分离得到含量较高的几个生物碱单体,具有一定的应用价值。如李佳莲采用pH- 区带精制逆流色谱法从钩吻总生物碱中一次性分离纯化出6个吲哚类生物碱单体(李佳莲.高速逆流色谱分离纯化麻黄、苍白秤钩风、钩吻和蟾酥中活性成分的研究[D].山东中医药大学,2013);Zhang Qiu-ping等通过高速逆流色谱法分离得到4个生物碱单体(Zhang Q P,Wang Z T,Chou G X.PreparativeSeparation of Four Alkaloids from Gelsemium elegans by High-speed Counter-current Chromatography [J].中草药(英文版),2015);刘浩等通过高速逆流色谱法分离得到钩吻素子(刘浩,沈洁,刘铭等,高速逆流色谱分离制备钩吻素子[J],中药新药与临床药理, 2013,24(2):197-200)和钩吻素甲(刘浩,沈洁,刘铭等,高速逆流色谱分离纯化钩吻中钩吻素甲[J],天然产物研究与开发,2013,25(4));沈洁等通过高速逆流色谱法分离得到钩吻素己和1-甲氧基钩吻碱(沈洁,苏燕评,许盈等,高速逆流色谱分离纯化钩吻中钩吻素己和1-甲氧基钩吻碱[J],中草药,2009,40(9): 1392-1395)。但上述提及的方法仅能分离少量钩吻碱单体。
发明内容
针对现有技术存在的缺陷,本发明的目的是在于提供一种利用高速逆流色谱法和制备液相色谱法相结合的方法从钩吻生物碱中快速高效分离制备多种钩吻生物碱单体的方法,该方法克服了现有技术中一般方法难以同时分离多种钩吻生物碱单体的缺陷。
为了实现上述技术目的,本发明提供了一种高速逆流色谱结合制备液相色谱从钩吻生物碱中分离钩吻生物碱单体的方法,该方法是将钩吻提取物采用高效液相色谱确定目标钩吻生物碱后,依次通过高速逆流色谱法初步分离和制备液相色谱法二次分离,分别收集各目标钩吻生物碱单体组分。
作为一个优选的方案,所述高效液相色谱法的条件:色谱柱:安捷伦 ZORBAX SB-C18色谱柱(4.6×250mm,5μm);流动相:0.1~0.3wt%磷酸-0.4~0.6wt%三乙胺混合水溶液和乙腈;采用梯度洗脱程序:0~65min,乙腈体积百分比由12%匀速升高至47%;高效液相色谱参数:流速0.8~1.2mL/min;柱温28~32℃;进样量15~25μL。最优选的流动相:0.2wt%磷酸-0.5wt%三乙胺混合水溶液和乙腈。最优选的高效液相色谱参数:流速1.0mL/min;柱温30℃;进样量20μL;检测波长为254nm。通过优化高效液相色谱参数可以尽量确定及获得更多的目标钩吻生物碱单体组分。
作为一个优选的方案,所述高速逆流色谱法的条件:采用的两相体系由 0.5~1.5wt%三乙胺水溶液、正己烷、乙酸乙酯、乙醇按体积比3~5:1~3:2~4:1~3 组成;高速逆流色谱仪参数:转速为800~900rpm/min,柱温箱为20~30℃,流速为1.5~2.5mL/min。较优选的高速逆流色谱法的条件:采用的两相体系由1wt%三乙胺水:正己烷:乙酸乙酯:乙醇溶液按体积比4:2:3:2组成。较优选的高速逆流色谱仪参数:转速为850rpm/min,柱温箱为25℃,流速为2mL/min,检测波长为254nm。
作为一个优选的方案,在高速逆流色谱进行初步分离过程中,依据高速逆流色谱图谱收集各目标钩吻生物碱单体组分:流分I:80~100min;流分II: 128~143min;流分III:187~230min;流分IV:249~298min;高速逆流色谱图谱采集至400min时,管柱内溶液上下相分离,上相为流分V,下相为流分VI。
作为一个优选的方案,所述制备液相色谱法的条件:色谱柱:UnitaryC18反相色谱柱(250mm×10mm,5μm);采用的流动相体系由0.4~0.6wt%三乙胺 -0.1~0.3wt%冰乙酸混合溶液和乙腈组成;采用梯度洗脱程序,依据制备液相色谱图谱收集各目标钩吻生物碱单体组分:流分I:0~50min:乙腈体积百分比由 13%匀速升高至50%,依次收集羟基钩吻素己和常绿钩吻碱;流分II:0~25min:乙腈体积百分比由22%匀速升高至30%;25~35min:乙腈体积百分比由30%匀速升高至60%;35~40min:乙腈体积百分比保持在60%,依次收集得到常绿钩吻碱和羟基胡蔓藤碱乙;流分III:0~10min:乙腈体积百分比由30%匀速升高至 40%;10~30min:乙腈体积百分比由40%匀速升高至50%;30~35min:乙腈体积百分比保持在50%,收集得到钩吻素子;流分IV:0~40min:乙腈体积百分比由25%匀速升高至60%;40~45min:乙腈体积比保持在60%,收集得到钩吻素甲;流分V:0~25min:乙腈体积百分比由30%匀速升高至45%;25~35min:乙腈体积百分比保持在45%;依次收集得到钩吻绿碱及11-羟基胡蔓藤碱乙;流分 VI:0~40min:乙腈体积百分比由30%匀速升高至50%;40~45min:乙腈体积百分比保持在50%,依次收集得到钩吻绿碱和胡蔓藤碱乙;制备液相色谱参数:流速为4~8mL/min。较优选的流动相体系由0.5wt%三乙胺-0.2wt%冰乙酸混合溶液和乙腈组成。较优选的制备液相色谱参数:流速为6mL/min,检测波长为254 nm。
本发明技术方案通过采用高速逆流色谱法结合制备液相色谱法可以从钩吻中分离得到8个生物碱单体,分别为羟基钩吻素己、常绿钩吻碱、羟基胡蔓藤碱乙、钩吻素子、钩吻素甲、钩吻绿碱、11-羟基胡蔓藤碱乙、胡蔓藤碱乙。
本发明的钩吻提取物是以新鲜钩吻鲜药材作为原料通过常规方法提取得到,具体如下:将4kg钩吻鲜药材洗净泥土,切成薄片,用4倍重量的95%乙醇冷浸提取3次,每次48h,合并提取液,通过旋转蒸发仪减压回收乙醇得浓缩液,往浓缩液中加入NaOH溶液调pH至10并静置分层,通过离心将上清液和沉淀分离,将上清液和沉淀分别用氯仿进行萃取,萃取至氯仿层无色,合并氯仿层,通过旋转蒸发仪减压回收氯仿将氯仿浓缩成浸膏,制得钩吻提取物,作为高速逆流色谱(HSCCC)的进样原料。
本发明通过制备液相色谱法分离的目标钩吻生物碱单体,经过55℃浓缩,冷冻干燥,获得高纯度的钩吻单体化合物粉末,最后利用高效液相色谱-质谱联用仪进行结构鉴定。
本发明采用高速逆流色谱法结合制备液相色谱法对钩吻提取物进行分离,具备可控性,且通过优化高效液相色谱条件,获得目标物,利用高速逆流色谱法结合制备液相色谱的方法,关键在于调整高速逆流色谱溶剂体系的具体配比参数,获得多个或单个流分;再通过制备液相色谱条件优化进行二次分离获得纯度更高的单个或多个单体成分,通过两者的结合高效分离出8个钩吻生物碱单体。
相对现有技术,本发明技术方案带来的有益技术效果:
(1)本发明技术方案高速逆流色谱法结合制备液相色谱法对钩吻提取物进行分离具有制备量大,周期短,高效等特点,高速逆流色谱法的使用不仅扩大了样品制备量,而且缩短了分离周期,在此基础上结合制备液相色谱法进行二次分离,同单独使用高速逆流色谱法进行二次分离相比,既缩短了分离时间,又可通过不断改变制备色谱条件收集多个不同极性的单体化合物,具备灵活性。
(2)本发明技术方案采用液-液色谱技术和固-液色谱技术联合应用,分离效率高,样品损耗少,同先前采用单一的高速逆流色谱法相比,可分离出更多的钩吻单体,钩吻生物碱的8个单体中只有一个单体为85.5%外,其余均在90%以上,甚至可达99%。
(3)本发明技术方案具有灵活性和可控性强的特点,采用多种色谱技术,实现自动化连接,利用高效液相色谱、高速逆流色谱和制备液相色谱三种色谱方法的结合。通过改变任意部分的溶剂体系可侧重分离不同数量的单体,且适用于不同极性样品的分离,具备自动化生产的条件,为药物生产和药理、毒理等科学研究提供基础。
附图说明
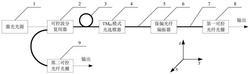
图1为钩吻提取物的高效液相色谱图;
图2为钩吻提取物的高速逆流色谱采集图谱;
图3和图4分别为流分I~流分VI的制备液相色谱图及分离目标生物碱单体的液相色谱图。
具体实施方式
以下结合具体实施例来对本发明作进一步说明,但本发明所要求保护的范围并不局限于具体实施例。
实施例1
一种高速逆流色谱法结合制备液相色谱从钩吻生物碱中分离钩吻生物碱单体的方法:
1、样品:钩吻生物碱,为广西柳州市柳城县龙头镇的钩吻鲜药材的溶剂提取物,依照传统的提取钩吻生物碱的方法(徐坚,梁崇真,钩吻总生物碱提取方法的比较[J],中国药学杂志.341-342(1988);张兰兰,硕士,第一军医大学,(2005);肖洒,硕士,湖南农业大学,(2016))。采用的化学试剂均为市售分析纯化学试剂。
2、仪器:分析型液相色谱仪(配有SPD-M20A检测器,LC-20AT输液泵, CBM-20A系统控制器,SIL-20A自动进样器,日本岛津公司);TBE-300A高速逆流色谱仪(内径1.6mm,柱容积280mL,中国上海同田生化技术有限公司);制备型LC8A高效液相色谱仪(日本岛津公司)。
3、方法:
3.1、钩吻提取物高效液相色谱法:对钩吻鲜药材进行醇提、碱化和萃取后,进行高效液相色谱检测以得到更多组分的目标物。检测条件如下:安捷伦 ZORBAX SB-C18色谱柱(4.6×250mm,5μm);流动相为超纯水(0.2%磷酸和 0.5%三乙胺)和乙腈,梯度洗脱程序0-65min,乙腈占比12%→47%(乙腈的体积百分比匀速增加);流速1.0mL/min;柱温30℃;进样量20μL;检测波长254 nm。钩吻提取物高效液相色谱图见图1。
3.2、高速逆流色谱溶剂体系筛选:钩吻提取物在高效液相色谱获得更多组分目标物后,摸索高速逆流色谱的两相体系;用正己烷、乙酸乙酯、乙醇、水、三乙胺按不同的体积比配置成溶液,移取3mL下相加入少量浸膏,用等体积上相进行萃取,在3.1条件下,分别测定萃前和萃后下相中目标峰面积,计算出各组分的分配系数(K值的计算:K=(S1-S2)/S2,式中K为各组分分配系数;S1 为萃取前的目标峰面积;S2为萃取后的目标峰面积)。选择K值适合的溶剂体系作为高速逆流色谱的溶剂体系并上机操作,图1中色谱峰较高的组分K值结果如表1所示。
表1图1中色谱峰较高的组分不同体系的K值
3.3、高速逆流色谱溶剂初步分离:按照1wt%三乙胺水:正己烷:乙酸乙酯: 乙醇=4:2:3:2组成体系配置溶液2200mL,静置分层过夜后将上相与下相分离,超声脱气10min后将上相溶剂以20mL/min的流速泵入主机管路,待上相溶剂充满管路后,高速逆流色谱进行分离,分离条件如下:转速850rpm/min,柱温箱 25℃,流速2mL/min,检测波长254nm。平衡后取钩吻提取物350mg,用20mL 下相溶解,进样并进行图谱采集,所得谱图见图2。
3.4、目标组分的收集:根据峰形收集流分I(80~100min)、流分II(128~143min)、流分III(187~230min)、流分IV(249~298min)流分。图谱采集至400min 时,停止仪器,用乙醇以20mL/min流速泵进管柱内,推出管柱内溶剂约320mL,推出液上、下相分离,上相为流分V,下相来流分VI。将6个不同的流分,分别减压浓缩至膏状后加入少量甲醇溶解,流分I(40mL)、流分II(20mL)、流分III(30mL)、流分IV(30mL)、流分V(30mL)、流分VI(40mL)过0.45μm滤膜,作为制备液相进样原料。
3.5、制备液相色谱法二次分离:将高速逆流色谱法初步分离收集到的各个组分通过减压浓缩回收有机试剂,用甲醇进行溶解,过膜,备用。利用制备型高效液相色谱对各个组分进行二次分离。制备型高效液相色谱分离条件如下:色谱柱型号:UnitaryC18反相色谱柱(250mm×10mm,5μm),流速为6mL/min,检测波长为254nm,根据峰形收集流出液,合并相同流分,各流分在55℃浓缩,冷冻干燥单体化合物粉末。6组流分的流动相梯度均为匀速变化,即流分I: 0-50min:乙腈13%→50%(乙腈体积百分比匀速增加,下同),收集得12.1mg 化合物1、12.3mg化合物2,HPLC面积归一法测定纯度分别为90.2%、98.9%;流分II:0-25min:乙腈22%→30%,25-35min:乙腈30%→60%,35-40min:乙腈60%→60%,收集得8.5mg化合物2、10.1mg化合物3,HPLC面积归一法测定纯度分别为98.5%、97.3%;流分III:0-10min:乙腈30%→40%,10-30min:乙腈40%→50%,30-35min:乙腈50%→50%,收集得50.5mg化合物4,HPLC面积归一法测定纯度为99.0%。流分IV:0-40min:乙腈25%→60%,40-45min:乙腈60%→60%,收集得32.2mg化合物5,HPLC面积归一法测定纯度为99.5%。流分V:0-25min:乙腈30%→45%,25-35min:乙腈45%→45%,22.0mg化合物6,12.5mg化合物7,HPLC面积归一法测定纯度分别为95.5%,96.8%,85.5%。流分VI:0-40min:乙腈30%→50%,40-45min:乙腈50%→50%,收集得28.5mg 化合物6,7.8mg化合物8,HPLC面积归一法测定纯度分别为95.53%,94.8%。流分I→VI的制备液相色谱图及分离化合物液相色谱图见图3和图4
3.6单体化合物结构鉴定
将上述得到的单体化合物粉末利用高效液相色谱-质谱联用仪,鉴定出单体化合物的结构。结构如下:化合物1:羟基钩吻素己;化合物2:常绿钩吻碱;化合物3:羟基胡蔓藤碱乙;化合物4:钩吻素子;化合物5:钩吻素甲;化合物6:钩吻绿碱;化合物7:11-羟基胡蔓藤碱乙;化合物8:胡蔓藤碱乙。
一种高速逆流色谱结合制备液相色谱从钩吻生物碱中分离钩吻生物碱单体的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



![芳基[a]吲哚[2,3-g]并喹嗪类化合物、其制备方法、药物组合物及其应用](https://www.zhichawang.com/images/ui/CN2012103473402/CN2012103473402.jpg)





动态评分
0.0