

IPC分类号 : B01J31/26,B01J37/34,C07C4/24,C07C4/00,C07C15/24,C10G1/06







专利摘要
一种微波法直接制备负载固体超强酸催化剂的方法,属于超强酸催化剂的制备方法。(1)将重量比为1:500的NaHCO3与碳纳米管充分混合搅拌,并置于微波反应器中焙烧,获得负载Na催化剂载体;(2)将负载Na催化剂载体放在微波辐射加热,逐滴加入五氯化锑(SbCl5)和三氟甲磺酸三甲基硅酯(F3CSO3Si(CH3)3)混合溶液,搅拌、浸渍,获得悬浮混合液;(3)过滤悬浮混合液,获得固体前驱体和混合溶液;(4)将固体前驱体放置于微波反应器中,微波辐射加热、焙烧,获得固体超强酸催化剂雏形;(5)以固体超强酸催化剂雏形为原料,按照步骤(2)、(3)和(4)操作1-5次,完成。优点:利用微波法制备的负载固体超强酸催化剂的催化加氢裂解活性高且具有较好的选择性。
说明书
技术领域
本发明涉及一种超强酸催化剂的制备方法,特别是一种微波法直接制备负载固体超强酸催化剂的方法。
背景技术
酸催化反应涉及到烃类裂解、重整、异构等石油炼制过程,还涉及到烯烃水合、烯烃聚合、芳烃烷基化、芳烃酰基化、醇酸酯化等石油化工和精细化工众多单元反应过程,可以说酸催化剂是一系列重要工业的基础。
目前,固体超强酸催化剂主要包括以下几类,分别是利用了异构化反应、烷基化反应、酯化反应、酰基化反应、低聚反、取代反应、缩醛和缩酮的单元反应,直接制备超强酸催化剂。
(1) 将SbF5、TaF5、BF3、SbF3-HF等一元或多元液体酸负载在适当的多孔氧化物、石墨、离子交换树脂等载体上;
(2) 将两种或两种以上的无机盐混合制得的固体超强酸,如AlCl3-CuCl、AlCl3-Ti2(SO4)3;
(3) 将树脂用氟改性制得全氟高分子材料Nafion-H;
(4) 将分子筛改性制得强酸型分子筛,如高酸性丝光沸石HM;
(5) 将含磷、钨、锗等元素的化合物经不同化学或物理过程制得杂多酸;
(6) 将两种或两种以上的金属氧化物焙烧制得的固体超强酸,如W03/TiO2-ZrO2;
(7) 将硫酸根负载于金属氧化物表面焙烧制得固体超强酸,如SO42-/MxOy;
(8) 负载酸与金属复合物协同超强酸催化剂,如在SO42-/ZrO2催化剂中在添加改善催化剂活性和寿命的组分(如Pt)等等。
上述超强酸催化剂制备复杂、时间长,催化剂的有效组分易于流失、热稳定性差,特别是无显著的芳环桥键催化加氢作用,催化效率低。
发明内容
本发明的目的是要提供一种微波法直接制备负载固体超强酸催化剂的方法,解决现在超强酸催化剂制备复杂、时间长,催化剂的有效组分易于流失、热稳定性差,特别是无显著的芳环桥键催化加氢作用,催化效率低的问题。
本发明的目的是这样实现的:制备方法是:
(1)将重量比为1:500的NaHCO3与碳纳米管充分混合搅拌,NaHCO3的重量浓度为1‰至1%;并将混合后的物料放置于300W至320W功率的微波反应器中,在氮气流中保持微波辐射加热的温度在200°C至220 °C,静止焙烧10分钟至20分钟后获得负载Na催化剂载体;
(2)在微波反应器中的烧瓶内放置10g至500g上述制备好的负载Na催化剂载体,在100W至120W功率微波照射下,在氮气流气氛中微波辐射加热的温度在30°C至50 °C,逐滴往烧瓶中加入总重量为100g至1000g、体积比为1:1至1:5的五氯化锑(SbCl5)和三氟甲磺酸三甲基硅酯(F3CSO3Si (CH3)3)混合溶液,体系充分搅拌、浸渍并保持10分钟至20分钟后获得浸渍SbCl5和F3CSO3Si (CH3)3混合溶液的超强酸催化剂固体前驱体的悬浮混合液;
(3)在氢气流保护下,借助附有有机膜的抽滤瓶过滤上述负载SbCl5和F3CSO3Si (CH3)3超强酸催化剂固体前驱体的悬浮混合液,获得浸渍SbCl5和F3CSO3Si (CH3)3混合液的超强酸催化剂的固体前驱体和剩余的SbCl5和F3CSO3Si (CH3)3混合溶液;获得的剩余SbCl5和F3CSO3Si (CH3)3混合溶液可作为步骤(2)中的混合浸渍剂循环使用;
(4)将上述获得10g至100g浸渍SbCl5和F3CSO3Si (CH3)3混合液的超强酸催化剂的固体前驱体放置于400W至500W功率的微波反应器中,在氢气流中保持微波辐射加热的温度在350°C至400 °C,静止焙烧60分钟至120分钟后获得固体超强酸催化剂雏形;
(5)以步骤(4)中多次获得的固体超强酸催化剂雏形为原料,按照步骤(2)、(3)和(4)步骤反复操作1-5次,完成了固体超强酸催化剂的全部制备过程,在氢气保护下用取出固体超强酸催化剂并密闭、备用。
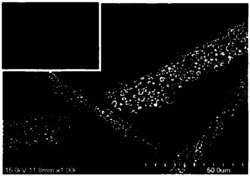
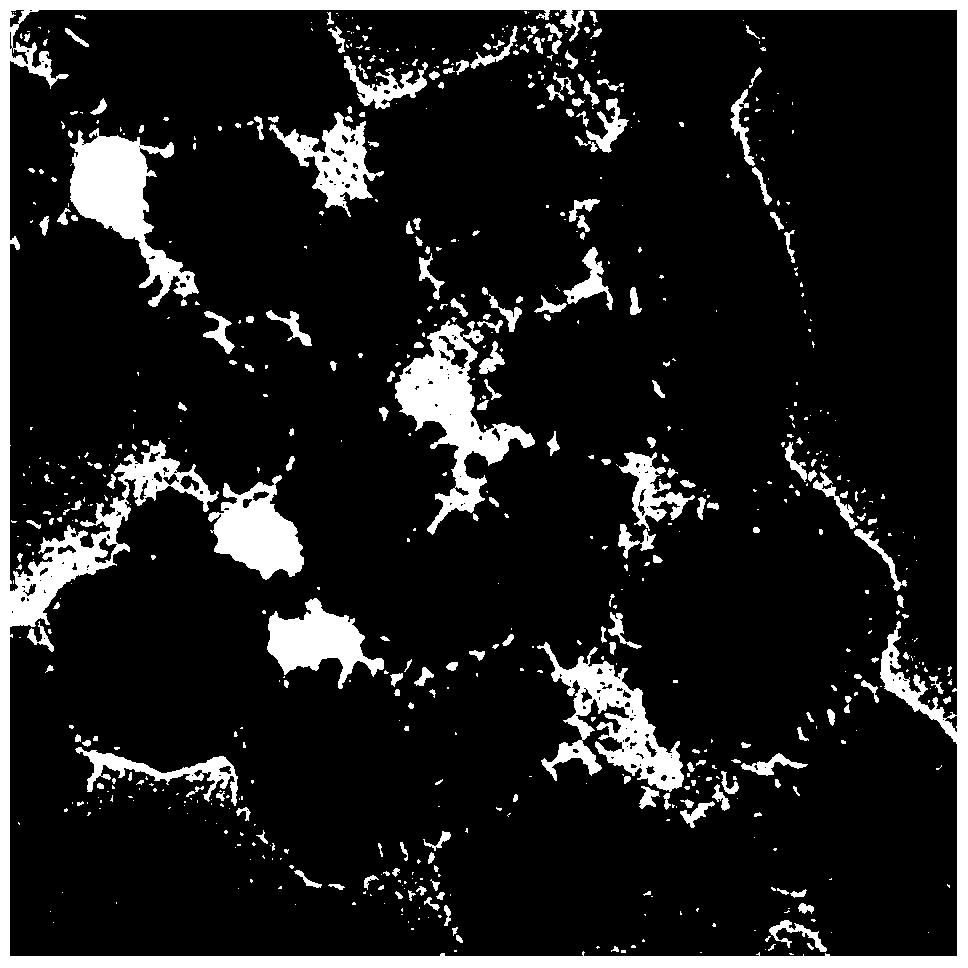
有益效果,由于采用了上述方案,通过TPD-NH3程序升温脱附图确定了上述几种方案中所制备的新型负载SbCl5和F3CSO3Si (CH3)3 /碳纳米管固体超强酸催化剂的表面酸强度Ho均小于-10.80,制备的固体酸催化剂属于固体超强酸催化剂范畴。实验结果表明该类催化剂对含有芳换环桥键样品的催化加氢裂解活性高且选择性好,实验过程中对设备无腐蚀、可以多次重复使用和再生、在150℃-350℃使用温度范围的催化效果最佳。采用全微波加热制备的新型载体固体超强酸催化剂具有适宜的比表面积、有效组分晶化速率快、晶粒均匀、边界清晰且更加均匀细小和致密孔径分布均匀且规整,高度分散在载体的内外表面上,有效阻止了负载晶体的催化剂粒子间的团聚效果,采用微波法制备固体酸催化剂有效缩短了制备催化剂的时间,改善催化剂表面的晶体类型,催化剂的热稳定性良好,工艺过程简单,效率高,达到了本发明的目的。
优点:利用微波法制备的新型负载SbCl5和F3CSO3Si (CH3)3 /碳纳米管固体超强酸催化剂可以促使芳换环桥键样品的催化加氢裂解活性高且具有较好的选择性,特别是用于煤及煤模型化合物桥键催化加氢裂解反应。
本发明方法制备超强酸催化剂时间短,催化剂的热稳定性好,催化剂的有效组分稳定,特别是适用于芳环桥键催化加氢作用,本催化剂的加氢转化效率和选择性显著提高。
具体实施方式
实施例1:负载固体超强酸催化剂的制备方法、操作条件以及催化剂的性能指标和催化效果:
1、催化剂的制备:
(1)将重量为1g、重量浓度为1%的NaHCO3与重量为500g的碳纳米管充分混合搅拌,并将混合后的物料放置于300W功率的微波反应器中,在氮气流中保持微波辐射加热的温度在220°C静止焙烧10分钟后获得负载Na催化剂载体。
(2)在微波反应器中的烧瓶内放置100g上述制备好的负载Na催化剂载体,在100W功率微波照射中,在氮气流中微波辐射加热的温度在50 °C条件下,逐滴往烧瓶中加入总重量为200g、体积比为1:2的SbCl5和F3CSO3Si (CH3)3混合溶液,体系充分搅拌、浸渍并保持20分钟后获得浸渍SbCl5和F3CSO3Si (CH3)3混合溶液的超强酸催化剂固体前驱体的悬浮混合液。
(3)在氢气流保护下,借助附有有机膜的抽滤瓶过滤上述负载SbCl5和F3CSO3Si (CH3)3超强酸催化剂固体前驱体的悬浮混合液,获得浸渍SbCl5和F3CSO3Si (CH3)3混合液的超强酸催化剂的固体前驱体和剩余的SbCl5和F3CSO3Si (CH3)3混合溶液。获得的剩余SbCl5和F3CSO3Si (CH3)3混合溶液可作为步骤(2)中的浸渍剂循环使用。
(4)将上述获得的浸渍SbCl5和F3CSO3Si (CH3)3混合液的超强酸催化剂的10g固体前驱体放置于500W功率的微波反应器中,在氢气流中保持微波辐射加热的温度在400°C,静止焙烧60分钟后获得固体超强酸催化剂雏形。
(5)以步骤(4)中10次获得的固体超强酸催化剂雏形为原料,按照步骤(2)、(3)和(4)步骤反复操作3-5次,完成了固体超强酸催化剂的全部制备过程,在氢气保护下用取出固体超强酸催化剂并密闭、备用。
2、催化剂的性能指标和催化效果:
负载固体超强酸催化剂中有效元素的质量含量分别为:1.6% O、4.5% F、0.1% Si、0.1% S、0.6% Cl、0.9% Sb,比表面积为487.6 m2 /g,总孔容为0.38 cm3 /g,平均孔径55.95 ?,催化剂超强酸的Ho酸度为- 11.20。在反应器中加入原料二萘甲烷1g,催化剂的绝对量为 0.4 g,初始压力为5MPa,保持反应温度在300 oC下范围内搅拌3h,二萘甲烷的转化率为98%。主要有效产物包括萘和甲基萘,两者单程的选择性均为100%。通过与传统方法热分解获得的催化剂比较,本方法制备的超强酸催化剂具有更高的桥键催化加氢裂解效果。
实施例2:负载固体超强酸催化剂的制备方法、操作条件以及催化剂的性能指标和催化效果:
1、催化剂的制备:
(1)将重量为1g、重量浓度为8‰的NaHCO3与重量为500g的碳纳米管充分混合搅拌,并将混合后的物料放置于300W功率的微波反应器中,在氮气流中保持微波辐射加热的温度在200°C静止焙烧20分钟后获得负载Na催化剂载体。
(2)在微波反应器中的烧瓶内放置200g上述制备好的负载Na催化剂载体,在100W功率微波照射中,在氮气流中微波辐射加热的温度在30 °C条件下,逐滴往烧瓶中加入总重量为500g、体积比为1:4的SbCl5和F3CSO3Si (CH3)3混合溶液,体系充分搅拌、浸渍并保持15分钟后获得浸渍SbCl5和F3CSO3Si (CH3)3混合溶液的超强酸催化剂固体前驱体的悬浮混合液。
(3)在氢气流保护下,借助附有有机膜的抽滤瓶过滤上述负载SbCl5和F3CSO3Si (CH3)3超强酸催化剂固体前驱体的悬浮混合液,获得浸渍SbCl5和F3CSO3Si (CH3)3混合液的超强酸催化剂的固体前驱体和剩余的SbCl5和F3CSO3Si (CH3)3混合溶液。获得的剩余SbCl5和F3CSO3Si (CH3)3混合溶液可作为步骤(2)中的浸渍剂循环使用。
(4)将上述获得的浸渍SbCl5和F3CSO3Si (CH3)3混合液的超强酸催化剂的50g固体前驱体放置于500W功率的微波反应器中,在氢气流中保持微波辐射加热的温度在350°C,静止焙烧100分钟后获得固体超强酸催化剂雏形。
(5)以步骤(4)中2次获得的固体超强酸催化剂雏形为原料,按照步骤(2)、(3)和(4)步骤反复操作3-5次,完成了固体超强酸催化剂的全部制备过程,在氢气保护下用取出固体超强酸催化剂并密闭、备用。
2、催化剂的性能指标和催化效果:
负载固体超强酸催化剂中有效元素的质量含量分别为:2.0% O、4.2% F、0.2% Si、0.1% S、0.3% Cl、0.6% Sb,比表面积为492.0m2 /g,总孔容为0.39 cm3 /g,平均孔径3.12 nm,催化剂超强酸的Ho酸度为- 10.89。在反应器中加入原料二萘甲烷1g,催化剂的绝对量为 0.6g,初始压力为5MPa,保持反应温度在290 oC下范围内搅拌3h,二萘甲烷的转化率为97%。主要有效产物包括萘和甲基萘,两者单程的选择性均为100%。通过与传统方法热分解获得的催化剂比较,本方法制备的超强酸催化剂具有更高的桥键催化加氢裂解效果。
实施例3:负载固体超强酸催化剂的制备方法、操作条件以及催化剂的性能指标和催化效果:
1、催化剂的制备:
(1)将重量为1g、重量浓度为4‰的NaHCO3与重量为500g的碳纳米管充分混合搅拌,并将混合后的物料放置于300W功率的微波反应器中,在氮气流中保持微波辐射加热的温度在200°C静止焙烧20分钟后获得负载Na催化剂载体。
(2)在微波反应器中的烧瓶内放置150g上述制备好的负载Na催化剂载体,在120W功率微波照射中,在氮气流中微波辐射加热的温度在40 °C条件下,逐滴往烧瓶中加入总重量为600g、体积比为1:1的SbCl5和F3CSO3Si (CH3)3混合溶液,体系充分搅拌、浸渍并保持20分钟后获得浸渍SbCl5和F3CSO3Si (CH3)3混合溶液的超强酸催化剂固体前驱体的悬浮混合液。
(3)在氢气流保护下,借助附有有机膜的抽滤瓶过滤上述负载SbCl5和F3CSO3Si (CH3)3超强酸催化剂固体前驱体的悬浮混合液,获得浸渍SbCl5和F3CSO3Si (CH3)3混合液的超强酸催化剂的固体前驱体和剩余的SbCl5和F3CSO3Si (CH3)3混合溶液。获得的剩余SbCl5和F3CSO3Si (CH3)3混合溶液可作为步骤(2)中的浸渍剂循环使用。
(4)将上述获得的浸渍SbCl5和F3CSO3Si (CH3)3混合液的超强酸催化剂的100g固体前驱体放置于500W功率的微波反应器中,在氢气流中保持微波辐射加热的温度在400°C,静止焙烧120分钟后获得固体超强酸催化剂雏形。
(5)以步骤(4)中1次获得的固体超强酸催化剂雏形为原料,按照步骤(2)、(3)和(4)步骤反复操作3-5次,完成了固体超强酸催化剂的全部制备过程,在氢气保护下用取出固体超强酸催化剂并密闭、备用。
2、催化剂的性能指标和催化效果:
固体超强酸催化剂中有效元素的质量含量分别为:1.7% O、4.0% F、0.2% Si、0.1% S、0.5% Cl、1.1% Sb,比表面积为477.6 m2 /g,总孔容为0.43 cm3 /g,平均孔径54.00 ?,催化剂超强酸的Ho酸度为- 11.4。在反应器中加入原料灵武次烟煤1g,催化剂的绝对量为 0.5 g,初始压力为5MPa,保持反应温度在280 oC下范围内搅拌4h,灵武次烟煤催化加氢的效率约为45%,主要有效产物包括萘和甲基萘,苯环类一元环产物、萘环类二元环产物及部分蒽和菲等其他三元环产物。通过与传统方法热分解获得的催化剂比较,本方法制备的超强酸催化剂具有更高的桥键催化加氢裂解效果。
一种微波法直接制备负载固体超强酸催化剂的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0