







专利摘要
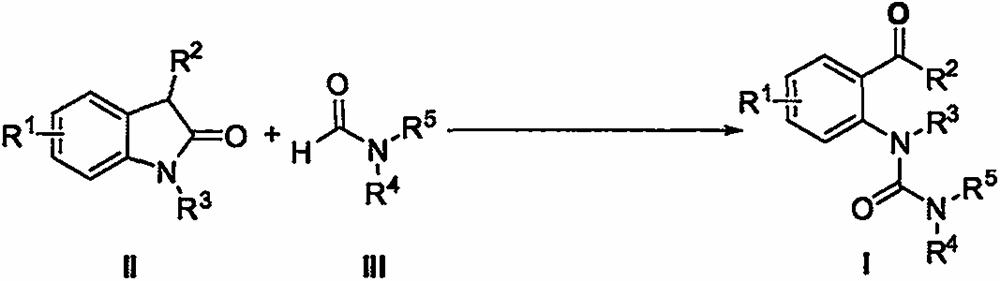
本发明属于有机合成领域,具体涉及一种不对称尿素衍生物的制备方法。式II的2‑吲哚酮类化合物与式III的甲酰胺类化合物在铜催化剂、过氧化叔丁醇为氧化剂的条件下,加热搅拌反应一段时间,得到式I所示的不对称尿素衍生物;
权利要求
1.一种不对称尿素衍生物的制备方法,其特征在于,式II的2-吲哚酮类化合物与式III的甲酰胺类化合物在铜催化剂、过氧化叔丁醇为氧化剂的条件下,加热搅拌反应一段时间,得到式I所示的不对称尿素衍生物;
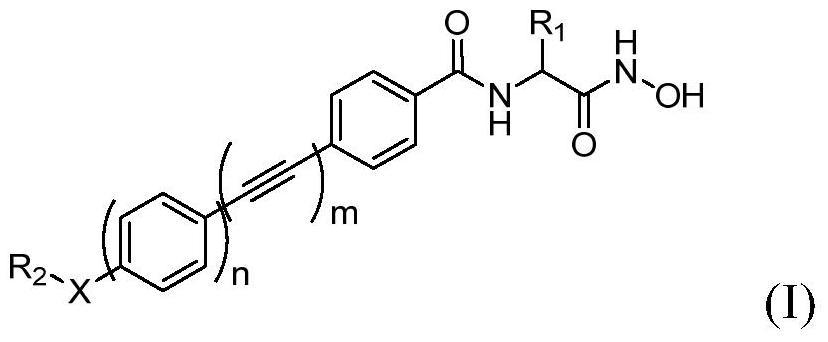
上述式I、式II、式III中,
R
所述的铜催化剂选自醋酸铜、氯化铜、溴化铜、碘化亚铜、溴化亚铜的一种或几种的混合物。
2.根据权利要求1所述的方法,其特征在于,所述的R
3.根据权利要求1-2任意一项所述的方法,其特征在于,所述方法的操作如下:向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II的2-吲哚酮类化合物和式III的甲酰胺类化合物,再加入铜催化剂和过氧叔丁醇;然后将反应器置于配备磁力搅拌的油浴锅中,加热搅拌反应,反应完全后,经后处理得到式I的不对称尿素衍生物。
4.根据权利要求3所述的方法,其特征在于,所述的加热搅拌反应的温度范围为80-120℃;所述反应的反应时间为0.5-3小时。
5.根据权利要求4所述的方法,其特征在于,所述的加热搅拌反应的温度为100℃,所述反应的反应时间为1-2小时。
6.根据权利要求1-2任意一项所述的方法,其特征在于,式II的2-吲哚酮类化合物、式III的甲酰胺类化合物、铜催化剂和过氧叔丁醇的投料摩尔比为1∶5-50∶0.05-0.3∶1-5。
7.根据权利要求6所述的方法,其特征在于,式II的2-吲哚酮类化合物、式III的甲酰胺类化合物、铜催化剂和过氧叔丁醇的投料摩尔比为1∶20∶0.2∶4。
8.根据权利要求3所述的方法,其特征在于,所述的后处理操作如下:反应完全后,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离得到式I的目标产物。
说明书
技术领域
本发明属于有机合成领域,具体涉及一种不对称尿素衍生物的制备方法。
背景技术
尿素衍生物是指含有尿素基本结构单元的一类有机化合物,即在尿素的氮原子上含有取代基的化合物,当两个氮原子上的取代基并不彼此呈对称的结构时,这类化合物被称之为不对称尿素衍生物。不对称尿素衍生物具有独特的结构特点和生理活性,广泛应用于生物医药和功能材料中。其已知的代表性化合物如下(可考现有技术文献1等:Coppercatalyzed oxidative coupling of amines with formamides:a new approach for thesynthesis of unsymmetrical urea derivatives,K.Rajender Reddy等,Chem.Commun.,2013,49,6686.):HIV-1蛋白酶抑制剂,NK1选择性拮抗剂,脂肪酸酰胺水解酶(FAAH)抑制剂,白血病药物,驱虫药(结构式见下式一)。
此外,专利文献2(JPH06211814A)则是公开了一种用于预防和/或治疗高血压的血管紧张素II拮抗剂,其中药效分子涉及如下通式的不对称尿素衍生物(参见式二);以及专利文献3(US200702600641A1)公开了一种用于治疗帕金森症的酒石酸匹莫范色林(Pimavanserin,Nuplazid)(式三)。
对于尿素衍生物的合成,传统的制备方法通常有如下几种:1)胺类物质与二氧化碳反应生成尿素衍生物;2)胺类物质与光气反应生成尿素衍生物;3)胺类物质与异氰酸酯反应生成尿素衍生物;4)胺类物质与CO在金属有机催化剂催化下反应生成尿素衍生物;5)胺类物质与尿素或者碳酸酯反应生成尿素衍生物。然而,这些方法使用的原料毒性巨大且价格昂贵,从而限制了这些方法的使用。
发明内容
本发明目的在于克服现有技术的不足,提供一种工艺简单、成本低、环境友好的制备不对称尿素衍生物的新方法。该方法以2-吲哚酮衍生物和甲酰胺类化合物为原料、在铜催化剂和过氧化叔丁醇为氧化剂的条件下进行反应,制备获得不对称尿素衍生物。
本发明上述目的是通过以下技术方案来实现的:
式II的2-吲哚酮类化合物与式III的甲酰胺类化合物在铜催化剂、过氧化叔丁醇为氧化剂的条件下,加热搅拌反应一段时间,得到式I所示的不对称尿素衍生物(式四)。
上述式I、式II、式III中,所述R1表示所连接苯环上的一个或多个取代基,选自氢、C1-C20的烷基、C1-C20的烷氧基、C1-C20的烷硫基、C6-C20的芳基、C3-C20的杂芳基、C3-C20的环烷基、硝基、卤素、-OH、-SH、-CN、-COOR6、-COR7、-OCOR8、-NR9R10;其中,R6、R7、R8、R9、R10各自独立地表示氢、C1-C20的烷基、C6-C20的芳基、C3-C20的杂芳基、C3-C20的环烷基中的任意一种或多种。
其中,上述各取代基中的烷基、芳基、杂芳基、环烷基部分可任选地被一个或多个选自C1-C6的烷基、C1-C6的烷氧基、卤素、-NO2、-CN、-OH、C6-C20的芳基、C3-C6的环烷基所取代。
优选地,所述R1表示所连接苯环上的一个或多个取代基,选自C1-C6的烷基、C1-C6的烷氧基、C1-C6的烷硫基、C6-C14的芳基、C3-C12的杂芳基、C3-C8的环烷基、硝基、卤素、-OH、-SH、-CN、-COOR6、-COR7、-OCOR8、-NR9R10;其中,R6、R7、R8、R9、R10各自独立地表示氢、C1-C6的烷基、C6-C12的芳基、C3-C12的杂芳基、C3-C8的环烷基中的任意一种或多种。且各取代基中的烷基、芳基、杂芳基、环烷基部分可任选地被一个或多个选自C1-C6的烷基、C1-C6的烷氧基、卤素、-NO2、-CN、-OH、C6-C12的芳基、C3-C6的环烷基所取代。
进一步优选地,所述C1-C6的烷基可以选自甲基、乙基、丙基、异丙基、丁基、正丁基、异丁基、叔丁基、戊基、异戊基、新戊基;所述C1-C6的烷氧基可以选自甲氧基、乙氧基、丙氧基、丁氧基;所述的C6-C12的芳基可以选自苯基、萘基、蒽基;所述的C3-C12的杂芳基可以选自噻吩基、咪唑基、吡啶基;所述的C3-C8的环烷基可以选自环丙基、环丁基、环己基;其中上述各基团可以任选地被一个或多个选自C1-C6的烷基、C1-C6的烷氧基、卤素、-NO2、-CN、-OH、C6-C12的芳基、C3-C6的环烷基所取代。
R2表示氢、C1-C6烷基、C3-C6环烷基、C5-C14芳基、C5-C14杂芳基。其中上述各R2基团可以任选地被一个或多个选自C1-C6的烷基、C1-C6的烷氧基、卤素、-NO2、-CN、-OH、C6-C12的芳基、C3-C6的环烷基所取代。
优选地,所述R2基团定义中C1-C6的烷基可以选自甲基、乙基、丙基、异丙基、丁基、正丁基、异丁基、叔丁基、戊基、异戊基、新戊基;所述C5-C14芳基优选为苯基或萘基;所述的C3-C6的环烷基选自环丙基、环丁基、环已基。
R3表示氢、叔丁氧羰基(Boc)、C1-C6烷基、C1-C6酰基、C3-C6环烷基、C5-C14芳基、C5-C14芳基-C1-C6烷基、C5-C14杂芳基。其中上述各R3基团可以任选地被一个或多个选自C1-C6的烷基、C1-C6的烷氧基、卤素、-NO2、-CN、-OH、C6-C12的芳基、C3-C6的环烷基所取代。
优选地,所述R3基团定义中C1-C6的烷基可以选自甲基、乙基、丙基、异丙基、丁基、正丁基、异丁基、叔丁基、戊基、异戊基、新戊基;所述C5-C14芳基优选为苯基;所述C5-C14芳基-C1-C6烷基优选为苄基。
R4和R5彼此独立地表示氢、C1-C6烷基、C3-C6环烷基、C5-C14芳基;或者R4和R5与所连接的氮原子一起,形成具有2-6个碳原子的并具有0-2个另外的N、O或S杂原子的环状结构。其中上述各R4和R5基团,以及所述R4和R5与所连接的氮原子一起形成的环状结构可以任选地被一个或多个选自C1-C6的烷基、C1-C6的烷氧基、卤素、-NO2、-CN、-OH、C6-C12的芳基、C3-C6的环烷基所取代。
优选地、R4和R5彼此独立地表示甲基、乙基、丙基、异丙基、正丁基、异丁基、叔丁基、戊基、异戊基、新戊基、苯基、苄基、或者R4和R5与所连接的氮原子一起形成吗啡啉基、哌啶基。
在本文中,如无特殊说明,所述杂芳基的杂原子应当理解为本领域常见的杂原子种类,例如可以选自O、S或N。
在前述式四的反应中,所述的铜催化剂选自醋酸铜、氯化铜、溴化铜、硫酸铜、碘化亚铜的一种或几种的混合物。
在前述式四的反应中,所述的反应不需要使用保护气氛,即在空气条件下进行。并且优选地,所述的反应不使用其它的有机溶剂。
在前述式四的反应中,所述的一定温度的温度范围为80-120℃,优选为100℃。所述反应的反应时间可以通过本领域常规技术手段例如GC-MS或TLC板进行监测,一般反应时间为0.5-3小时,优选为1-2小时。
本发明中,所述反应的典型操作如下:
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II的2-吲哚酮类化合物和式III的甲酰胺类化合物,再加入铜催化剂和过氧化叔丁醇;然后将反应器置于配备磁力搅拌的油浴锅中,加热搅拌反应一段时间,反应完全后,经后处理得到式I的不对称尿素衍生物。
其中,式II的2-吲哚酮类化合物、式III的甲酰胺类化合物、铜催化剂和过氧化叔丁醇的投料摩尔比为1∶5-50∶0.05-0.3∶1-5;优选地,式II的2-吲哚酮类化合物、式III的甲酰胺类化合物、铜催化剂和过氧化叔丁醇的投料摩尔比为1∶20∶0.2∶4。
所述的后处理操作如下:反应完全后,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到式I的目标产物。
本发明的有益效果如下:
1.本发明提供的方法首次利用自由基串联反应策略由2-吲哚酮和甲酰胺类化合物构建不对称尿素衍生物。
2.本发明的方法首次发现了无碱体系中2-吲哚酮开环的新途径,并且能进一步与甲酰胺类化合物发生自由基偶联反应。
3.本发明的方法经过C-O偶联,开环反应,C-N偶联的反应过程,在切断一个C-C键和C-N键的同时,构建了一个新的C=O键和C-N键。
4.本发明的方法反应使用廉价低毒的铜催化剂,过氧化叔丁醇在反应中既作为氧化剂,又作为氧源。
具体实施方式
以下结合具体实施例,对本发明进行进一步详细的描述。
实施例1-15反应条件优化
如反应式五所示,以式II-1的3-苯基-2-吲哚酮化合物和式III-1的N,N-二甲基甲酰胺为反应原料,制备式I-1的不对称尿素衍生物,探讨了不同反应条件对于合成工艺优化结果的影响,选择出其中具有代表性的实施例1-15,结果如表一所示:
其中实施例1的典型操作如下:
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-1的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-1的目标化合物,产率74%。
表一:
其中,实施例2-15的具体操作除上述表一所列的变量与实施例1不相同之外,其余操作均与实施例1相同。
由实施例1-15可以看出,本发明的自由基串联反应必须在过氧化叔丁醇作为氧化剂的条件下进行(实施例2),以及不使用催化剂Cu(OAc)2的条件下反应收率下降明显(实施例3);不同的铜催化剂中Cu(OAc)2的催化效果最佳(实施例4-7),其投料量为2-吲哚酮类化合物投料量的20mmol%为最好(实施例8-9);其他的氧化剂例如K2S2O8等代替过氧化叔丁醇则使得反应无法进行和/或反应收率明显降低(实施例10-12),而过氧化叔丁醇的投料量进一步增加则不能进一步提高目标产物的收率(实施例13);反应温度的升高或降低均会对目标产物的收率带来一定的影响(实施例14-15)。
由上述实施例1-15可以看出,最佳的反应条件为实施例1的反应条件,即催化剂选择Cu(OAc)2、氧化剂选择过氧化叔丁醇、反应温度设定为100℃。在获得最佳反应条件的基础上,发明人进一步在该最佳反应条件下,选择不同取代基的反应原料以制备各种不对称尿素衍生物。
实施例16
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-1的3-苯基-2-吲哚酮(0.3mmol)和式III-2的N,N-二乙基甲酰胺(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-2的目标化合物,产率60%。
实施例17
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-1的3-苯基-2-吲哚酮(0.3mmol)和式III-3的甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-3的目标化合物,产率63%。
实施例18
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-2的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-4的目标化合物,产率61%。
实施例19
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-3的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-5的目标化合物,产率75%。
实施例20
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-4的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-6的目标化合物,产率77%。
实施例21
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-5的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-7的目标化合物,产率62%。
实施例22
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-6的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到棕色固体的式I-8的目标化合物,产率58%。
实施例23
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-7的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到白色固体的式I-9的目标化合物,产率45%。
实施例24
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-8的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到无色油状的式I-10的目标化合物,产率56%。
实施例25
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-9的2-吲哚酮类化合物(0.3mmol)和式III-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-11的目标化合物,产率63%。
实施例26
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-10的2-吲哚酮类化合物(0.3mmol)和式m-1的N,N-二甲基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-12的目标化合物,产率61%。
实施例27
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-11的2-吲哚酮类化合物(0.3mmol)和式III-2的N,N-二乙基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到棕色固体的式I-13的目标化合物,产率48%。
实施例28
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-12的2-吲哚酮类化合物(0.3mmol)和式III-2的N,N-二乙基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-14的目标化合物,产率60%。
实施例29
向Schlenk封管反应器中加入一颗磁力搅拌子,加入式II-13的2-吲哚酮类化合物(0.3mmol)和式III-2的N,N-二乙基甲酰胺化合物(0.5mL),再加入Cu(OAc)2(0.06mmol)和过氧化叔丁醇(1.2mmol);然后将反应器置于配备磁力搅拌的油浴锅中,于100℃加热搅拌反应1小时,经TLC监测反应完全,将反应液用乙酸乙酯稀释,用饱和食盐水洗涤,水相用乙酸乙酯萃取,合并有机相,用无水硫酸钠干燥,过滤,减压条件下浓缩,并将获得的浓缩物通过硅胶柱层析分离(乙酸乙酯/正己烷)得到黄色油状的式I-15的目标化合物,产率50%。
以上所述实施例仅为本发明的优选实施例,而并非本发明可行实施的穷举。对于本领域技术人员而言,在不背离本发明原理和精神的前提下,对其所作出的任何显而易见的改动,都应当被认为包含在本发明的权利要求保护范围之内。
一种不对称尿素衍生物的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0