







专利摘要
本发明提出了一种合成喹啉衍生物的方法,其是在反应容器中按摩尔比1:1:1.2~2依次加入芳香族胺、吸电子炔烃、酮,按1mmol芳香族胺加入2~4mL的比例加入溶剂,接着加入催化剂Cu(OTf)2(三氟甲磺酸铜)和添加剂HOTf(三氟甲磺酸),加入量分别为芳香族胺摩尔量的0.8%~5%和1.8%~10%,在100℃~120℃油浴条件下反应10~24h,冷却至室温,加水,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,得到产品喹啉衍生物。本发明具有反应底物廉价,产率高、选择性好、易分离纯化、污染少,步骤简单的特点。
权利要求
1.一种合成喹啉衍生物的方法,其特征是包括以下方法步骤:
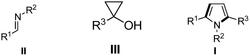
在反应容器中按摩尔比1:1:1.2~2依次加入芳香族胺Ⅰ、吸电子炔烃Ⅱ、酮Ⅲ,按1mmol芳香族胺加入2~4 mL的比例加入溶剂,接着加入催化剂三氟甲磺酸铜Cu(OTf)2和添加剂三氟甲磺酸HOTf,加入量分别为芳香族胺摩尔量的0.8%~5%和1.8%~10%,在100℃~120℃油浴条件下反应10~ 24h,冷却至室温,加水,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,得到产品喹啉衍生物,合成方法的反应通式表示如下:
该方法合成的喹啉衍生物其化学结构通式如下:
其中,R1 为H、C1~C6 链状烷基、C1~C6 链状烷氧基、NO2、OH、F、Cl或者Br;酮Ⅲ为2-乙酰基吡啶、邻氟苯乙酮、间氟苯乙酮、对氟苯乙酮、邻氯苯乙酮、间氯苯乙酮、对氯苯乙酮、邻溴苯乙酮、间溴苯乙酮、对溴苯乙酮、邻甲氧基苯乙酮、间甲氧基苯乙酮、对甲氧基苯乙酮、邻甲基苯乙酮、间甲基苯乙酮、对甲基苯乙酮、2-乙酰基噻吩或者2-乙酰基吡咯。
2.根据权利要求1所述的一种合成喹啉衍生物的方法,其特征是所述的芳香族胺Ⅰ为苯胺、邻氟苯胺、间氟苯胺、对氟苯胺、邻氯苯胺、间氯苯胺、对氯苯胺、邻溴苯胺、间溴苯胺、对溴苯胺、邻硝基苯胺、间硝基苯胺、对硝基苯胺、邻甲氧基苯胺、间甲氧基苯胺、对甲氧基苯胺、邻甲基苯胺、间甲基苯胺或对甲基苯胺。
3.根据权利要求1所述的一种合成喹啉衍生物的方法,其特征是所述的溶剂为甲苯、乙腈或者THF。
4.根据权利要求1所述的一种合成喹啉衍生物的方法,其特征是所述的柱层析条件为:300~400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:2~1:10。
说明书
技术领域
本发明涉及喹啉衍生物,具体涉及一种合成喹啉衍生物的方法。
背景技术
喹啉及其衍生物是一类重要有机杂环化合物,在自然界中广泛存在,被广泛地应用于药物筛选、化学分析、染料工业等领域。合成喹啉衍生物方法有很多,主要有:Skraup-Doebner-Miller合成法,Friedlander-Pfitzinge-Combes合成法,Bischler-Napieralski合成法(Tetrahedron.Lett,2000,41,531-533;Org.lett,2004,6,3965-3968;Eur.J.Org.Chem,2008,2693-2696及J.Org.Chem,2003,68,3966-3975),其中使用最为广泛的是Skraup-Doebner-Miller法。喹啉类化合物的合成方法一直是人们研究的热点(参见:G.R.Humphrey,J.T.Kuethe,Chem,Rev,2006,106,2875),目前工业上合成喹啉最有代表性的方法有Skraup合成法,其以芳胺、浓硫酸、甘油与温和氧化剂一起加热,制得喹啉衍生物。在反应过程中,甘油在高温下受浓硫酸作用脱水为丙烯醛,再与苯胺缩合为二氢喹啉,最后经氧化得到喹啉,常用硝基苯或砷酸作为催化剂法,但是该反应要在浓硫酸中高温进行。Combes合成法,以芳香胺与β-二酮在酸性环境中缩合为喹啉环。该法以芳胺与1,3-二羰基化合物缩合得到β-氨基-烯酮,后者在浓硫酸的作用下进行环合,得到喹啉衍生物。但是,当芳胺环上有吸电子基团存在时,会使苯环上电子云密度降低,从而不利于亲电取代反应的进行。当1,3-二碳基化合物(R1-CO-CH2CO-R2)中的R1与R2不同时,则第一步的缩合反应有两种可能,会生成两种β-氨基-烯酮,环合产物即为含有两种同分异构体的混合物。
上世纪八十年代开始报道了过渡金属催化的端炔化合物与邻位带有卤素的苯胺先发生偶联反应生成邻氨基炔,再环化制备喹啉的方法(参见:(a)Müller,T.E.;Beller,M.Chem.Rev.1998,98,675;(b)Roundhill,D.M.Chem.Rev.1992,92,1.(c)Bryndza,H.E.;Tam,W.Chem.Rev.1988,88,1163),是合成喹啉类衍生物的重要方法(参见:(a)Hartung,C.G.;Breindl,C.;Tillack,A.;Beller,M.Tetrahedron 2000,56,5157;(b)Kawatsura,M.;Hartwig,J.F.J.Am.Chem.Soc.2000,122,9546;(c)Beller,M.;Trauthwein,H.;Eichberger,M.;Breindl,C.;Müller,T.E.Eur.J.Inorg.Chem.1999,11.21;(d)Trauthwein,H.;Tillack,A.;Beller,M.Chem.Commun.1999,2029;(e)Beller,M.;Trauthwein,H.;Eichberger,M.;Breindl,C.;Herwig,J.;Müller,T.E.;Thiel,O.R.Chem.Eur.J.1999,5,1306;(f)Beller,M.;Trauthwein,H.;Eichberger,M.;Breindl,C.;Müller,T.E.;Zapf,A.J.Organomet.Chem.1998,566,277),已成功的运用到天然产物和仿生药物的合成上(参见:Z.Z.Shi,C.Zhang,S.Li,D.l.Pan,S.T.Ding,Y.X.Cui,N.Jiao,Angew.Chem.Int.Ed.2009,48,4572)。
中国专利CN104151235A公开了一种喹啉衍生物制备方法,是利用三氟甲磺酸银催化取代基的苯胺和烯酮或烯醛衍生物合成喹啉衍生物,本专利操作简单,不仅能够适用于大量的官能团,而且产率高,产物单一,便于分离和提纯、安全廉价、污染小。但是本专利仍然存在一些缺点:不能够制备2,4位上为吸电子酯基的喹啉衍生物。
目前的合成方法存在很多缺点:主要是反应条件苛刻,反应温度高,有的需要高温高压,分离困难,反应的底物限制性较强,因此一种方法合成取代基的喹啉衍生物很有限。另外,利用金属催化过程中,催化剂的活性有限,这些缺点造成制备过程的操作难度增加,危害操作人员健康,环境污染严重。现有合成喹啉衍生物的方法还普遍存在:需要活泼的反应底物、反应差率低、反应时间较长、副产物多且难处理以及反应的形式过于单一(导致所合成的产物有很大的局限性)等缺点。鉴于此,研发新颖的喹啉衍生物的制备方法显得尤为重要。
发明内容
针对现有合成喹啉衍生物所存在的缺陷,本发明所要解决的技术问题是提供一种合成喹啉衍生物的方法,该方法操作简单,产率高,产物易分离和提纯,且污染少。
为解决上述技术问题,本发明所采取的技术方案是:一种合成喹啉衍生物的方法,包括以下方法步骤:
在反应容器中按摩尔比1:1:1.2~2依次加入芳香族胺Ⅰ、吸电子炔烃Ⅱ、酮Ⅲ,按1mmol芳香族胺加入2~4mL的比例加入溶剂,接着加入催化剂Cu(OTf)2(三氟甲磺酸铜)和添加剂HOTf(三氟甲磺酸),加入量分别为芳香族胺摩尔量的0.8%~5%和1.8%~10%,在100℃~120℃油浴条件下反应10~24h,冷却至室温,加水,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,得到产品喹啉衍生物,合成方法的反应通式表示如下:
该方法合成的喹啉衍生物其化学结构通式如下:
其中,R1为H、C1~C6链状烷基、C1~C6链状烷氧基、NO2、OH、F、Cl或者Br;R2为H、甲酸酯基、C1~C6链状烷基或者苯基;R3为吡啶基、C3~C6烷基、环烷基、噻吩基、芳基。
所述的芳香族胺Ⅰ为苯胺、邻氟苯胺、间氟苯胺、对氟苯胺、邻氯苯胺、间氯苯胺、对氯苯胺、邻溴苯胺、间溴苯胺、对溴苯胺、邻硝基苯胺、间硝基苯胺、对硝基苯胺、邻甲氧基苯胺、间甲氧基苯胺、对甲氧基苯胺、邻甲基苯胺、间甲基苯胺、对甲基苯胺、邻三氟苯胺、间三氟苯胺或者对三氟苯胺。
所述的吸电子炔烃Ⅱ为丁炔二酸二甲酯、丁炔二酸二乙酯、苯丙炔酸甲酯、苯丙炔酸乙酯、丙炔酸甲酯、丁炔酸甲酯、丙炔酸乙酯或者丁炔酸乙酯。
所述的酮Ⅲ为2-乙酰基吡啶、邻氟苯乙酮、间氟苯乙酮、对氟苯乙酮、邻氯苯乙酮、间氯苯乙酮、对氯苯乙酮、邻溴苯乙酮、间溴苯乙酮、对溴苯乙酮、邻甲氧基苯乙酮、间甲氧基苯乙酮、对甲氧基苯乙酮、邻甲基苯乙酮、间甲基苯乙酮、对甲基苯乙酮、邻三氟苯乙酮、间三氟苯乙酮、对三氟苯乙酮、1-戊酮、1-己酮、1-庚酮、2-乙酰基噻吩或者2-乙酰基吡咯。
所述的溶剂为甲苯、已腈或者THF。
所述的柱层析条件为:300~400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:2~1:10。
本发明采用上述技术方案所设计的一种合成喹啉衍生物的方法,其有益效果为:
1.本发明酸用量少,减少了环境污染;
2.本发明反应底物简单,来源广泛,适合各种官能团取代的苯胺(包括邻位大位阻的官能团取代的苯胺),立体效应对反应的影响小;
3.本发明部分产物,如:反应通式Ⅳ位上为吡啶取代基,可以制备功能性材料的主核;
4.本发明反应底物廉价,产率高、选择性好、易分离纯化、污染少,步骤简单,可以省略官能团的保护和去保护合成步骤,目标产物喹啉衍生物广泛适用于有机化学反应的配体、药物中间体和光电材料方面。
附图说明
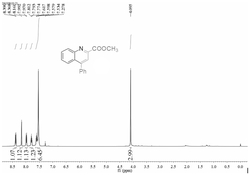
图1表示化合物4-(吡啶-2-基)喹啉-2-羧酸甲酯的
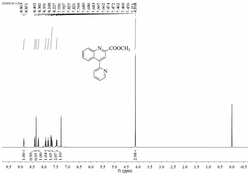
图2表示化合物4-(吡啶-2-基)喹啉-2-羧酸甲酯的
具体实施方式
下面结合实施例,对本发明一种合成喹啉衍生物的方法作详细说明。
实施例1
一种4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入苯胺1.0mmol(93mg),丁炔二酸二甲酯1.0mmol(142.1mg),2-乙酰基吡啶1.2mmol(145.2mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在100℃油浴中反应10h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品240.3mg,产率91%,纯度99.9%。
其中,所述的4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构通式为
实施例2
一种6-甲基-4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入对甲基苯胺1.0mmol(107mg),丁炔二酸二甲酯1.0mmol(142.1mg),2-乙酰基吡啶1.2mmol(145.2mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf0.01mmol(1.5mg),溶剂已腈2mL,在100℃油浴中反应10h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品267.0mg,产率96%,纯度99.9%。
其中,所述的6-甲基-4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构通式为:
实施例3
一种6-甲氧基-4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入对甲氧基苯胺1.0mmol(123.1mg),丁炔二酸二甲酯1.0mmol(142.1mg),2-乙酰基吡啶1.2mmol(145.2mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在100℃油浴中反应10h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品273.5mg,产率93%,纯度99.9%。
其中,所述的6-甲氧基-4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构
通式为
实施例4
一种6-氟-4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入对氟苯胺1.0mmol(111mg),丁炔二酸二甲酯1.0mmol(142.1mg),2-乙酰基吡啶1.2mmol(145.2mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应24h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:2,得到白色固体产品234.1mg,产率83%,纯度99.9%。
其中,所述的6-氟-4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构通
式为:
实施例5
一种6-氯-4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入对氯苯胺1.0mmol(127mg),丁炔二酸二甲酯1.0mmol(142.1mg),苯乙炔1.2mmol(122.4mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应12h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:3,得到白色固体产品241.4mg,产率82%,纯度99.9%。δppm:δ8.87(d,J=4.0Hz,1H),8.28-8.33(m,3H),7.92-7.96(t,1H),7.75(d,J=9.2Hz,1H),7.69(d,J=8.0Hz,1H),7.46-7.49(m,1H),4.10(s,3H);
其中,所述的6-氯-4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构通
式为:
实施例6
一种6-溴-4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入对溴苯胺1.0mmol(171mg),丁炔二酸二甲酯1.0mmol(142.1mg),苯乙炔1.2mmol(122.4mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应12h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:3,得到白色固体产品287.3mg,产率84%,纯度99.9%。
其中,所述的6-溴-4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构通式为:
实施例7
一种7-甲基-4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入间甲基苯胺1.0mmol(107mg),丁炔二酸二甲酯1.0mmol(142.1mg),苯乙炔1.2mmol(122.4mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应12h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品253.1mg,产率91%,纯度99.9%。
其中,所述的7-甲基-4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构通式为:
实施例8
一种7-甲氧基-4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入间甲氧基苯胺1.0mmol(123.1mg),丁炔二酸二甲酯1.0mmol(142.1mg),苯乙炔1.2mmol(122.4mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应12h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品267.6mg,产率91%,纯度99.9%。
其中,所述的7-甲氧基-4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构通式为:
实施例9
一种8-甲基-4-(吡啶-2-基)喹啉-2-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入邻甲基苯胺1.0mmol(1107mg),丁炔二酸二甲酯1.0mmol(142.1mg),苯乙炔1.2mmol(122.4mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应12h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品230.8mg,产率83%,纯度99.9%。
其中,所述的8-甲基-4-(吡啶-2-基)喹啉-2-羧酸甲酯化学结构通式为:
实施例10
一种7-甲氧基-2,4-二苯基喹啉的合成方法,包括以下步骤:
在反应容器中依次加入间甲氧基苯胺1.0mmol(123.1mg),苯丙炔酸甲酯1.0mmol(160.2mg),苯乙炔1.2mmol(122.4mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在100℃油浴中反应8h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品296.4mg,产率95%,纯度99.9%。
其中,所述的7-甲氧基-2,4-二苯基喹啉化学结构通式为:
实施例11
一种7-三氟甲基-2,4-二苯基喹啉的合成方法,包括以下步骤:
在反应容器中依次加入间三氟甲基苯胺1.0mmol(161.1mg),苯丙炔酸甲酯1.0mmol(160.2mg),苯乙炔1.2mmol(122.4mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应24h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品287.1mg,产率82%,纯度99.9%。
其中,所述的7-三氟甲基-2,4-二苯基喹啉化学结构通式为:
实施例12
一种2-甲基-6-甲氧基-4-苯基喹啉的合成方法,包括以下步骤:
在反应容器中依次加入对甲氧基苯胺1.0mmol(123.1mg),丁炔酸甲酯1.0mmol(98.1mg),苯乙炔1.2mmol(122.4mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在100℃油浴中反应8h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:10,得到白色固体产品182.6mg,产率73%,纯度99.9%。
其中,所述的2-甲基-6-甲氧基-4-苯基喹啉化学结构通式为:
实施例13
一种7,8,9,10-四氢菲啶-6-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入苯胺1.0mmol(93mg),丁炔二酸二甲酯1.0mmol(142.1mg),环己酮1.5mmol(147.2mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应12h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:10,得到白色固体产品226.6mg,产率94%,纯度99.9%。
其中,所述的7,8,9,10-四氢菲啶-6-羧酸甲酯化学结构通式为:
实施例14
一种2-甲基-7,8,9,10-四氢菲啶-6-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入对甲基苯胺1.0mmol(107mg),丁炔二酸二甲酯1.0mmol(142.1mg),环己酮1.5mmol(147.2mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf 0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应12h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:5,得到白色固体产品247.4mg,产率97%,纯度99.9%。
其中,所述的2-甲基-7,8,9,10-四氢菲啶-6-羧酸甲酯化学结构通式为:
实施例15
一种2-三氟甲基-7,8,9,10-四氢菲啶-6-羧酸甲酯的合成方法,包括以下步骤:
在反应容器中依次加入对三氟甲基苯胺1.0mmol(161mg),丁炔二酸二甲酯1.0mmol(142.1mg),环己酮1.5mmol(147.2mg),催化剂Cu(OTf)20.005mmol(1.8mg),HOTf0.01mmol(1.5mg),溶剂已腈2mL,在120℃油浴中反应12h,冷却至室温,加水5mL,用乙酸乙酯萃取三次,合并有机层,减压浓缩,产品经过柱层析纯化,300-400目硅胶柱,洗脱剂为乙酸乙酯和石油醚的混合物,两者体积比为1:10,得到白色固体产品268.9mg,产率87%,纯度99.9%。
其中,所述的2-三氟甲基-7,8,9,10-四氢菲啶-6-羧酸甲酯化学结构通式为:
本发明发展了一种利用三氟甲磺酸银催化取代基的苯胺、吸电子炔烃和酮合成喹啉衍生物的方法,该方法不仅适用于供电子取代的苯胺,对于吸电子的苯胺也有较好的产率。本方法是一种操作简便、安全便宜、高效的制备取代喹啉类化合物的新型方法。与现有技术相比,此方法不仅能够适用于大量的官能团,而且操作简单、产率高,产物单一,便于分离和提纯、安全廉价、污染小。
一种合成喹啉衍生物的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)











动态评分
0.0