

IPC分类号 : C07C235/82,C07C231/12,A01N37/42,A01P3/00







专利摘要
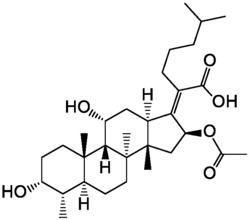
一类具有杀菌活性的多取代2‑羟基‑1,2‑二氢化萘酮衍生物。通式(I)为具有杀菌活性的羟基二氢化萘酮衍生物,具体为:式中R1为4‑氯苯基、4‑溴苯基,R1为叔丁基、正丁基、环己基。上述化合物对意大利青霉菌、指状青霉菌和稻瘟菌具有较好的抑制活性,可用作杀菌剂。
权利要求
1.一类多取代2-羟基-1,2-二氢化萘酮衍生物的制备方法,其特征在于,具有通式(I)表达的结构:
其中,R
化合物季膦盐羧酸(III)与酮醛和异腈按摩尔比1:1:1,在30℃下反应,反应溶剂为二氯甲烷,反应24小时后,溶剂换成甲苯,温度升高到50℃,加入三乙胺,摩尔用量为季膦盐羧酸(III)的两倍,继续反应2-5小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到通式(I)的目标化合物。
2.权利要求1所述的多取代2-羟基-1,2-二氢化萘酮衍生物的制备方法,其特征在于,所述的酮醛为对氯苯乙酮醛、对溴苯乙酮醛。
3.权利要求1所述的多取代2-羟基-1,2-二氢化萘酮衍生物的制备方法,其特征在于,所述的异腈为叔丁基异腈、环己基异腈或正丁基异腈。
4.权利要求1-3任一项所制备得到的多取代2-羟基-1,2-二氢化萘酮衍生物在制备指状青霉菌Penicillium digitatum、意大利青霉菌Penicillium italicum及稻瘟菌Magnaporthe grisea的杀菌剂上的应用。
说明书
技术领域
本发明涉及2-羟基-1,2-二氢化萘酮衍生物,以及它作为杀菌剂的有效成分的应用。
背景技术
民以食为天,粮食的安全生产关系到国计民生,是生死攸关的大问题。粮食的生产离不开农药,安全、低毒、高效的农药是粮食丰收的强有力保障,杀菌剂是农药的重要组成部分,在农药的发展史中扮演着重要角色。但是,不断变异的细菌和真菌以及各种不同条件下对杀菌剂使用限制的需求,研究开发出更多具有更好抑菌活性的新型杀菌剂,是促进杀菌剂发展的关键。鉴于氢化萘酮类衍生物具有良好的杀菌活性,尤其是在氢化萘酮环上含有羟基的此类化合物。研究新型的羟基氢化萘酮类衍生物的制备及其杀菌活性具有重要的意义。
发明内容
本发明的主要目的在于探索杀菌性良好的化合物,提供具有杀菌活性的2-羟基-1,2-二氢化萘酮衍生物。
本发明提出了2-羟基-1,2-二氢化萘酮衍生物(I):
其中,取代基R
本发明提供上述式(I)的化合物对意大利青霉菌、指状青霉菌和稻瘟菌具有较好的抑制活性,因而可作为杀菌剂的有效成分。
本发明合成方法是将季膦盐羧酸与酮醛、异腈在碱的存在下以“一锅法”反应制备,式(II)中的R
所述方法包括以下步骤:
向反应瓶中加入甲苯,称取通式(II)所示的化合物加入到反应瓶中,加入两倍当量的三乙胺作为碱,于50℃下搅拌2-5个小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到通式(I)的目标化合物。
合成原料的(II)的实验步骤:
所述化合物季膦盐羧酸(III)与酮醛(IV)和异腈(V)按摩尔比1:1:1,在30℃下反应,反应溶剂为二氯甲烷,反应24小时后,溶剂换成甲苯,加入三乙胺,用量为溴带羧酸(III)的1.2倍。反应在50℃下搅拌2-5小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到通式(II)的目标化合物。
具体实施方式
下面结合实施例来进一步说明本发明(I)式中化合物的制备和应用效果。
仪器及试剂:
熔点用X4型熔点仪(北京第三光学仪器厂生产)测定,温度计未经校正;
实施例1
的制备
向50mL烧瓶中加入季膦盐羧酸(III)(1mmol)、对氯苯乙酮醛(IV)(1mmol)和叔丁基异腈(V)(1mmol),在30℃下反应,反应溶剂为二氯甲烷,反应24小时后溶剂换成甲苯,加入三乙胺(2mmol),继续在50℃反应下搅拌3小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到0.313g白色晶体,产率85%。
元素分析:实测值 C% 68.43 H% 5.39 N% 3.87
计算值 C% 68.20 H% 5.45 N% 3.79
MS(EI,70eV):m/z(%)=369(1)[M+],325(6),270(100),241(56).
实施例2
的制备
向50mL烧瓶中加入季膦盐羧酸(III)(1mmol)、对氯苯乙酮醛(IV)(1mmol)和环己基异腈(V)(1mmol),在30℃下反应,反应溶剂为二氯甲烷,反应24小时后溶剂换成甲苯,加入三乙胺(2mmol),继续在50℃反应下搅拌3小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到0.329g白色晶体,产率83%。
元素分析:实测值 C% 69.97 H% 5.41 N% 3.70
计算值 C% 69.78 H% 5.60 N% 3.54
MS(EI,70eV):m/z(%)=395(4)[M+],351(19),270(100).
实施例3
的制备
向50mL烧瓶中加入季膦盐羧酸(III)(1mmol)、对氯苯乙酮醛(IV)(1mmol)和正丁基异腈(V)(1mmol),在30℃下反应,反应溶剂为二氯甲烷,反应24小时后溶剂换成甲苯,加入三乙胺(2mmol),继续在50℃反应下搅拌3小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到0.280g白色晶体,产率76%。
元素分析:实测值 C% 65.33 H% 5.61 N% 3.86
计算值 C% 68.20 H% 5.45 N% 3.79
MS(EI,70eV):m/z(%)=369(1)[M+],325(3),270(100).
实施例4
的制备
向50mL烧瓶中加入季膦盐羧酸(III)(1mmol)、对溴苯乙酮醛(IV)(1mmol)和叔丁基异腈(V)(1mmol),在30℃下反应,反应溶剂为二氯甲烷,反应24小时后溶剂换成甲苯,加入三乙胺(2mmol),继续在50℃反应下搅拌3小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到0.323g白色晶体,产率78%。
元素分析:实测值 C% 61.13 H% 4.59 N% 3.57
计算值 C% 60.88 H% 4.87 N% 3.38
MS(EI,70eV):m/z(%)=413(1)[M+],315(3),245(100).
实施例5
的制备
向50mL烧瓶中加入季膦盐羧酸(III)(1mmol)、对溴苯乙酮醛(IV)(1mmol)和环己基异腈(V)(1mmol),在30℃下反应,反应溶剂为二氯甲烷,反应24小时后溶剂换成甲苯,加入三乙胺(2mmol),继续在50℃反应下搅拌3小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到0.361g白色晶体,产率82%。
元素分析:实测值 C% 62.91 H% 4.77 N% 3.31
计算值 C% 62.74 H% 5.04 N% 3.18
MS(EI,70eV):m/z(%)=439(8)[M+],395(18),314(100),286(38),256(64).
实施例6
的制备
向50mL烧瓶中加入季膦盐羧酸(III)(1mmol)、对溴苯乙酮醛(IV)(1mmol)和正丁基异腈(V)(1mmol),在30℃下反应,反应溶剂为二氯甲烷,反应24小时后溶剂换成甲苯,加入三乙胺(2mmol),继续在50℃反应下搅拌3小时,反应完成后,反应液在减压下脱去溶剂,残留物柱层析得到0.302g白色晶体,产率73%。
元素分析:实测值 C% 60.97 H% 4.98 N% 3.52
计算值 C% 60.88 H% 4.87 N% 3.38
MS(EI,70eV):m/z(%)=413(1)[M+],371(13),314(100),286(44).
实施例7
杀菌活性实验(含毒介质法)
药液浓度100ppm,用5mm打孔器取菌种琼脂片,菌丝面朝下接种要含有待测药品的PDA培养基上,置于圆形培养基的正中心,切不要滑动菌种琼脂片,以免污染培养基。每个待测样品接种三个,以不含药品但含有相同浓度DMSO的培养基为对空白照,放置在生化培养箱内于25℃下培养3~5天后,测定培养基上的菌落的直径。通过和上述空白对照组的比较来观察待测样品对菌丝生长的影响,计算待测样品在100mg/L下对菌落生长的抑制率。抑制率(%)=[(空白对照菌落直径-待测样品菌落直径)/(空白菌落直径-打孔器直径)]×100%。表1为部分化合物(I)的测定结果。
表1:化合物(I)的抑菌活性测试结果
从上述表1可以看出,本发明的式(I)所表示的化合物对指状青霉菌(Penicilliumdigitatum)、意大利青霉菌(Penicillium italicum)及稻瘟菌(Magnaporthe grisea)具有较好的抑制活性。其中以化合物(I)-6效果最好。
本发明的化合物作为杀菌剂使用时,可将本发明的化合物与其它植保上允许的载体或稀释剂混合,借此将其调制成通常使用的各种剂型,如混剂、颗粒剂、水乳剂等来使用,也可以与其它农药如杀菌剂、杀虫剂、除草剂及植物生长调节剂混合使用或同时并用。
具有杀菌活性的多取代2-羟基-1,2-二氢化萘酮及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。









动态评分
0.0