








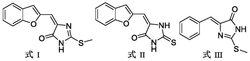
专利摘要
本发明属于有机化工技术领域,特别是一种作为杀菌剂的三唑吡咯烷酮类化合物,所述杀菌剂化学结构式为:其中,取代基R1和R2为苯基或对溴苯基或对氯苯基或对氟苯基或对甲基苯基及其他共轭体系,取代基位置、个数以及共轭位置不固定。合成方法为以一类三唑酰胺酮类化合物在催化剂作用下,经过分子内脱水缩合闭环反应,得到一类新型三唑吡咯烷酮类化合物。本发明提供了一类步骤少、产率高的合成新方法以合成一类新型三唑吡咯烷酮,又因产物其表现出良好的抑菌活性,有利于作为杀菌剂的应用。
权利要求
1.一种三唑吡咯烷酮类杀菌剂,其特征在于,所述杀菌剂的化学结构式为:
其中,取代基R
2.合成权利要求1所述的三唑吡咯烷酮类杀菌剂的方法,其特征在于,合成路径如下:
具体包括以下步骤:
(1)在反应瓶中配制混合溶剂,混合溶剂由乙醇与水按一定比例混合而成;
(2)向所述步骤(1)中的混合溶剂中加入化合物1,碳酸钾,将反应体系置于0~60℃下搅拌反应0.2~2小时,直至体系中出现大量固体;
所述化合物1为三唑酰氨酮;
(3)取所述步骤(2)的反应液TLC监测,反应完毕后,抽滤,剩余物经干燥即得到化合物2,即可完成三唑吡咯烷酮类杀菌剂的合成。
3.根据权利要求2的方法,其特征在于:所述步骤(1)配制混合溶剂使用的乙醇与水的体积比为1:1。
4.根据权利要求2的方法,其特征在于:所述步骤(2)化合物1与碳酸钾投料质量比为2:3。
5.根据权利要求2的方法,其特征在于:所述步骤(2)反应温度为0~60℃,反应时间为0.2~2小时。
6.权利要求2-5任意一项制备得到的三唑吡咯烷酮类杀菌剂在抑制稻瘟菌、指状青霉菌、意大利青霉菌、水稻纹枯菌上的应用。
7.权利要求6所述的应用,其特征在于,4-(4-氯苯基)-1-(4-氟苯基)-3-(1H-1,2,4-三唑)-1H-吡咯-2(5H)酮在抑制青霉菌上的应用,其结构式为:
8.权利要求6所述的应用,其特征在于,4-(4-氯苯基)-3-(1H-1,2,4-三唑)-1-(4-(三氟甲基)苯基)-1H-吡咯-2(5H)酮在抑制青霉菌上的应用,其结构式为:
说明书
技术领域
本发明涉及有机化工技术领域,特别是一种三唑吡咯烷酮类杀菌剂及其合成方法。
背景技术
粮食供应不足的问题一直以来都是人类面对的巨大挑战,尤其在非洲等农业不发达地区。而由各种细菌和真菌引起的农作物病害造成的粮食减产,带来的经济损失非常巨大。为了应对不断变异的细菌和真菌,以及各种不同条件下对杀菌剂使用限制的需求,研究开发出更多具有更好抑菌活性的新型杀菌剂,是促进杀菌剂发展的关键。目前商品化的三唑类杀菌剂主要特点为:大多数杀菌剂的药效团三唑环都连接在碳链上,且分子结构中含有酰胺键的杀菌剂有很多,但三唑类杀菌剂分子结构中含有酰胺键的却很少见报道。目前商业化的杀菌剂存在抑菌活性不够理想、半抑制浓度(IC50)较高等问题。
发明内容
本发明的主要目的在于提供一种三唑吡咯烷酮类杀菌剂及其合成方法。
本发明的技术方案如下:
一种三唑吡咯烷酮类杀菌剂,所述杀菌剂化学结构式为:
其中,取代基R1和R2为苯基或对溴苯基或对氯苯基或对氟苯基或对甲基苯基及其他共轭体系,取代基位置、个数以及共轭位置不固定。
合成所述的三唑吡咯烷酮类杀菌剂的方法,所述方法包括以下合成路径:
所述方法包括以下步骤:
1)在反应瓶中配制混合溶剂,混合溶剂由乙醇与水按一定比例混合而成;
2)向所述步骤1)中的混合溶剂中加入化合物1,碳酸钾,将反应体系置于0~60℃下搅拌反应0.2~2小时,直至体系中出现大量固体;
所述化合物1为三唑酰氨酮类衍生物;
3)取所述步骤2)的反应液TLC监测,反应完毕后,抽滤,剩余物经干燥即得到化合物2;
完成三唑吡咯烷酮类杀菌剂的合成。
所述步骤1)配制混合溶剂使用的乙醇与水的体积比为1:1。按此比例配制的混合溶剂,其溶解性较好,有利于反应的进行。
所述步骤2)化合物1与碳酸钾投料比为2:3。按此比例加入碳酸钾,则反应快速且平稳进行。碳酸钾加入量过少则反应缓慢进行或不反应,过多则导致反应剧烈,副产物增多。
所述步骤2)反应温度为0~60℃,高于此温度区间则反应加剧,副产物增多;低于此温度区间则反应时间延长或不反应。反应时间为0.2~2小时,少于0.2小时则反应不充分。
本发明有益效果如下:
1、本发明为以三唑酰胺酮衍生物在催化剂作用下经分子内缩合反应,将吡咯环引入三唑类化合物,得到的三唑吡咯烷酮类杀菌剂,其抑菌活性较高,半抑制浓度(IC50)较低,适合作为杀菌剂应用在各种不同环境条件下;
2、本发明合成一类与传统商业化杀菌剂不同的新型三唑吡咯烷酮类杀菌剂,提供了一种制备成本低、操作简单且反应效率高的合成新方法。
3、本发明合成了一类三唑吡咯烷酮类杀菌剂,此类化合物在抑制稻瘟菌、指状青霉菌、意大利青霉菌、水稻纹枯菌等菌类方面有较广泛的研究与应用。
具体实施方式
下面结合实施例来进一步说明本发明,但本发明要求保护的范围并不局限于实施例表述的范围。
实施例1
一种合成1-(4-溴苯基)-4-苯基-3-(1H-1,2,4-三唑)-1H-吡咯-2(5H)酮的方法,包括以下实验步骤:
称取N-(4-溴苯基)-N-(2-甲基苄基)-2-(1H-1,2,4-三唑)乙酰胺1(0.80g,2mmol)和碳酸钾(0.42g,3mmol)加入到10mL乙醇和水(V乙醇:V水=1:1)的混合溶剂中,于30℃下电磁搅拌,1小时后体系中出现大量固体,TLC监测,反应完毕后抽滤、干燥即得三唑吡咯烷酮2:
产率:87%
实施例2
一种合成4-(4-氯苯基)-1-(4-氟苯基)-3-(1H-1,2,4-三唑)-1H-吡咯-2(5H)酮的方法,包括以下实验步骤:
称取N-(2-(4-氯苯基)-2-氧代乙基)-N-(4-氟苯基)-2-(1H-1,2,4-三唑)乙酰胺1(0.75g,2mmol)和碳酸钾(0.42g,3mmol)加入到10mL乙醇和水(V乙醇:V水=1:1)的混合溶剂中,于40℃下电磁搅拌,1小时后体系中出现大量固体,TLC监测,反应完毕后抽滤、干燥即得三唑吡咯烷酮2:
产率:83%
实施例3
一种合成4-(4-氯苯基)-1-(对甲苯基)-3-(1H-1,2,4-三唑)-1H-吡咯-2(5H)酮的方法,包括以下实验步骤:
称取N-(2-(4-氯苯基)-2-氧代乙基)-N-(对甲苯基)-2-(1H-1,2,4-三唑)乙酰胺1(0.74g,2mmol)和碳酸钾(0.42g,3mmol)加入到10mL乙醇和水(V乙醇:V水=1:1)的混合溶剂中,于40℃下电磁搅拌,2小时后体系中出现大量固体,TLC监测,反应完毕后抽滤、干燥即得三唑吡咯烷酮2:
产率:79%
实施例4
一种合成4-(4-氟苯基)-1-(对甲苯基)-3-(1H-1,2,4-三唑)-1H-吡咯-2(5H)酮的方法,包括以下实验步骤:
称取N-(2-(4-氟苯基)-2-氧代乙基)-N-(对甲苯基)-2-(1H-1,2,4-三唑)乙酰胺1(0.70g,2mmol)和碳酸钾(0.42g,3mmol)加入到10mL乙醇和水(V乙醇:V水=1:1)的混合溶剂中,于40℃下电磁搅拌,2小时后体系中出现大量固体,TLC监测,反应完毕后抽滤、干燥即得三唑吡咯烷酮2:
产率:76%
实施例5
一种合成1-(4-氯苯基)-4-(4-氟苯基)-3-(1H-1,2,4-三唑)-1H-吡咯-2(5H)酮的方法,包括以下实验步骤:
称取N-(4-氯苯基)-N-(2-(4-氟苯基)-2-氧代乙基)-2-(1H-1,2,4-三唑)乙酰胺1(0.75g,2mmol)和碳酸钾(0.42g,3mmol)加入到10mL乙醇和水(V乙醇:V水=1:1)的混合溶剂中,于30℃下电磁搅拌,0.5小时后体系中出现大量固体,TLC监测,反应完毕后抽滤、干燥即得三唑吡咯烷酮2:
产率:71%
实施例6
一种合成4-(4-氯苯基)-3-(1H-1,2,4-三唑)-1-(4-(三氟甲基)苯基)-1H-吡咯-2(5H)酮的方法,包括以下实验步骤:
称取N-(4-氯苯基)-N-(2-(4-氟苯基)-2-氧代乙基)-2-(1H-1,2,4-三唑)乙酰胺1(0.75g,2mmol)和碳酸钾(0.42g,3mmol)加入到10mL乙醇和水(V乙醇:V水=1:1)的混合溶剂中,于30℃下电磁搅拌,1小时后体系中出现大量固体,TLC监测,反应完毕后抽滤、干燥即得三唑吡咯烷酮2:
产率:77%
实施例7
一种合成1-(4-氟苯基)-4-苯基-3-(1H-1,2,4-三唑)-1H-吡咯-2(5H)酮的方法,包括以下实验步骤:
称取N-(4-氟苯基)-N-(2-甲基苄基)-2-(1H-1,2,4-三唑)乙酰胺1(0.68g,2mmol)和碳酸钾(0.42g,3mmol)加入到10mL乙醇和水(V乙醇:V水=1:1)的混合溶剂中,于50℃下电磁搅拌,1.5小时后体系中出现大量固体,TLC监测,反应完毕后抽滤、干燥即得三唑吡咯烷酮2:
产率:75%
上述的实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本申请中的实施例及实施例中的特征在不冲突的情况下,可以相互任意组合。本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围。即在此范围内的等同替换改进,也在本发明的保护范围之内。
化合物波普性质:
2a:1-(4-bromophenyl)-4-phenyl-3-(1H-1,2,4-triazol-1-yl)-1H-pyrrol-2(5H)-one
White solid,mp 213-214℃;
2b:4-(4-chlorophenyl)-1-(4-fluorophenyl)-3-(1H-1,2,4-triazol-1-yl)-1H-pyrrol-2(5H)-one
White solid,mp 197-198℃;
2c:4-(4-chlorophenyl)-1-(p-tolyl)-3-(1H-1,2,4-triazol-1-yl)-1H-pyrrol-2(5H)-one
Yellow solid,mp 215-216℃;
2d:4-(4-fluorophenyl)-1-(p-tolyl)-3-(1H-1,2,4-triazol-1-yl)-1H-pyrrol-2(5H)-one
Light yellow solid,mp 176-177℃;
2e:1-(4-chlorophenyl)-4-(4-fluorophenyl)-3-(1H-1,2,4-triazol-1-yl)-1H-pyrrol-2(5H)-one
White solid,mp 182-182℃;
2f:4-(4-chlorophenyl)-3-(1H-1,2,4-triazol-1-yl)-1-(4-(trifluoromethyl)phenyl)-1H-pyrrol-2(5H)-one
Light yellow solid,mp 217-219℃;
2g:1-(4-fluorophenyl)-4-phenyl-3-(1H-1,2,4-triazol-1-yl)-1H-pyrrol-2(5H)-one
White solid,mp 188-189℃;
目标化合物三唑吡咯烷酮2的生物活性测试
设备和材料:
台式压力高温蒸汽灭菌箱(YXQ.DY型),感量万分之一的电子天平(SartoriusBT25S),无菌操作台(志诚,ZHJH-C1109B),接种针,玻璃棒,刻度尺,打火机,封口膜、三角瓶(75mL),量筒(20mL),微量进样器(100μL,200μL,500μL,1000μL),培养皿(直径6cm),霉菌培养箱(MJX-250型),细纱布,酒精灯,直径5mm打孔器。
PDA粉(青岛海洋化工),蒸馏水,DMSO,吐温-80,小麦赤霉菌(Gibberellasaubinetii),稻瘟菌(Magnaporthegrisea),指状青霉菌(Penicilliumdigitatum),意大利青霉菌(Penicillium italicum),水稻纹枯菌(Rhizoctonia solani)。
实验方法(采用含毒介质法)
PDA培养基的制备
称取PDA粉46g,加入蒸馏水1000mL,摇匀配置成溶液,用棉纱布盖上瓶口。把此培养基放到高压灭菌锅内采用湿热法于125℃下灭菌0.5h。
试样的配制(50mg/L)
用分析天平称取2~3mg待测样品放于已消毒的样品管中,加入几滴DMSO溶解该样品,加入适量蒸馏水和一滴吐温-80,配置成均匀的浓度为1000mg/L的溶液,取此溶液1000μL,迅速加入到9.0mL热的(40~50℃)培养基内,摇匀即配置成待测样品浓度为100mg/L的培养基,水平放置在无菌操作台内紫外杀菌15min。
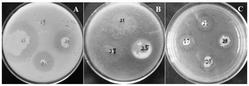
抑菌活性测定
用直径5mm打孔器取菌种琼脂片,菌丝面朝下接种要含有待测药品的PDA培养基上,置于圆形培养基的正中心,切不要滑动菌种琼脂片,以免污染培养基。每个待测样品接种三个,以不含药品但含有相同浓度DMSO的培养基为对空白照,以含相同浓度的商品化药物烯唑醇、三唑酮的作为对照,放置在生化培养箱内于25℃下培养3~5天后,测定培养基上的菌落的直径。通过和上述空白对照组以及商品化药物对照组的比较来观察待测样品对菌丝生长的影响,计算待测样品在100mg/L下对菌落生长的抑制率。
抑制率(%)=[(空白对照菌落直径-待测样品菌落直径)/(空白菌落直径-打孔器直径)]×100%
目标化合物三唑吡咯烷酮2的抑菌活性测试结果
表1:化合物2的抑菌活性测试结果
注:化合物浓度均为100mg/L;数据为三次重复的平均值。
从上述表1中我们可以看出,该系列化合物三唑吡咯烷酮2在100mg/L下对意大利青霉菌、指状青霉菌、水稻纹枯菌和稻瘟菌均表现出了一定的抑制活性,个别化合物抑菌活性达到了较高水平,如化合物2b对指状青霉菌的抑菌率达到了98%,对水稻纹枯菌的抑菌率也达到了94%。
一种三唑吡咯烷酮类杀菌剂,合成方法及其应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。










动态评分
0.0