

IPC分类号 : C07J43/00,C07F15/00,A61K31/555,A61P35/00




专利摘要
本发明属于药物合成领域,具体涉及一种孕酮受体靶向氮杂环卡宾中间体和钌类配合物及其制备方法与应用,通过合成式(Ⅰ)所示孕酮受体靶向氮杂环卡宾中间体进一步合成了式(Ⅱ)所示孕酮受体靶向钌类配合物,该配合物具有对GSH(谷胱甘肽)特异性响应性和对孕酮受体(PR)的靶向性;细胞实验显示其对孕酮受体阳性肿瘤细胞具有较高的细胞毒性和靶向性;动物实验也表明,该配合物对孕酮受体阳性肿瘤具有较高的靶向性及抗肿瘤活性,而且本发明提供的孕酮受体靶向钌类配合物的制备方法简单,产物易于提纯,该配合物可以作为靶向于PR受体的抗乳腺癌的药物。
权利要求
1.一种孕酮受体靶向氮杂环卡宾中间体,其特征在于,所述中间体具有式(Ⅰ)所示结构:
其中,R表示的为
2.一种如权利要求1所述的中间体的制备方法,其特征在于,包括如下步骤:
取
3.一种式(Ⅱ)所示的孕酮受体靶向钌类配合物的制备方法,其特征在于,包括如下步骤:
室温避光条件下,将权利要求2所述方法所制得的式(Ⅰ)所示的所述孕酮受体靶向氮杂环卡宾中间体溶于配位反应溶剂中,加入Ag
还包括半制备HPLC分离提纯步骤,其中,固定相为C18色谱柱,流动相A为含有0.1%TFA的去离子水,流动相B为含有0.1%TFA的乙腈,采用梯度洗脱收集产物,淋洗程序设定为:起始-3min时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B;
所述配合物具有式(Ⅱ)所示结构:
(Ⅱ)
其中,R表示的为
说明书
技术领域
本发明属于药物合成领域,具体涉及一种孕酮受体靶向氮杂环卡宾中间体和钌类配合物及其制备方法与应用。
背景技术
乳腺癌是当前威胁女性健康的主要疾病之一,发病率占全身各种恶性肿瘤发病率的7-10%,在女性肿瘤中仅次于子宫瘤,成为危害全球妇女健康的重大原因。辅助化疗是当前治疗乳腺癌的主要方法之一,可以降低乳腺癌原发肿瘤的分期和复发转移率,提高手术率和总生存率。然而,目前临床上的化疗药物对晚期患者效果不理想,仅限于缓解病情,并不能治愈,而且由于多数临床药物缺乏靶向性,长期服用会增加子宫内膜癌发生风险。
分子生物学的发展和对发病机制从细胞、分子水平的深入认识,使得肿瘤治疗进入了一个新的分子靶向治疗的时代。目前靶向递送抗癌药物到特定的细胞在药物研发领域有很大的潜力,将生物分子和抗癌基团相结合,以生物分子为靶向分子,携带药物基团定向进入肿瘤细胞,是提高药物抗肿瘤活性的良好方法。一方面结合后的药物可以具有生物分子的生理特性,如靶向特定作用位点,从而提高药物的靶向性,降低其毒副作用;另一方面可以增加细胞对药物的摄取,延长药物在细胞内的保留时间,从而提高药物活性。此外,在对乳腺癌的研究中,科学家发现孕酮受体(PR)在多种乳腺癌中都过度表达,孕酮受体可以作为治疗乳腺癌的一种重要靶点。
近年来,越来越多的研究表明钌化合物具有比铂类配合物更低的毒性以及较高的抗癌活性,成为研究热点。例如,中国专利文献CN106478734A公开了一种具有抗癌活性的吡唑官能团化的氮杂环卡宾钌化合物及其制备方法和应用,结构通式为[LRu(p-cymene)Cl]
其中,R1和R2独立地选自C1~C5烷基、取代或者未取代的苄基、取代或者未取代的苯基;所述的苄基或苯基上的取代基选自一个或多个C1~C5烷基、卤素、三氟甲基。
申请人前期工作中,也设计合成了一系列具有较高抗肿瘤活性的氮杂环卡宾钌类配合物,但是由于其缺乏靶向性,毒副作用较大,限制了其进一步的应用。
综上,研究利用具有较高的抗肿瘤活性的钌类配合物,并且结合分子靶向技术提高其对乳腺癌肿瘤细胞的选择性,以获得抗肿瘤效果较好、对正常细胞的毒副作用较小、制备方法较简便的孕酮受体靶向钌类配合物具有重要意义。
发明内容
为此,本发明提出一种孕酮受体靶向氮杂环卡宾中间体及其制备方法,并提供了一种孕酮受体靶向钌类配合物,并进而提供制备方法与应用。
为解决上述技术问题,本发明本发明提供了一种孕酮受体靶向氮杂环卡宾中间体,其具有式(Ⅰ)所示结构:
其中,R表示的为
本发明还提供了所述孕酮受体靶向氮杂环卡宾中间体的制备方法,
取 炔孕酮溶于聚合反应溶剂中,加入亚铜类催化剂发生加成聚合反应,分离纯化,即得式(Ⅰ)所示的孕酮受体靶向氮杂环卡宾中间体。
进一步优选地,包括如下步骤:取 、炔孕酮和亚铜类催化剂溶于聚合反应溶剂中,于37-60℃下反应8-20h,分离纯化,即得式(Ⅰ)所示的孕酮受体靶向氮杂环卡宾中间体,其中, 炔孕酮和亚铜类催化剂的摩尔比为1:(1-2):(1-4)。
进一步优选地, 炔孕酮、亚铜类催化剂和聚合反应溶剂的摩尔比为:1:(1-1.2):(1-2)。
进一步优选地,所述亚铜类催化剂为CuSO4·5H2O与抗坏血酸钠、氧化亚铜与醋酸、醋酸亚铜、碘化亚铜与四丁基醋酸铵、六氟磷酸四乙腈铜(I)中的一种。
进一步优选地,所述聚合反应溶剂为甲醇、乙醇、DMF、乙腈中的一种或者几种和水的混合物。
本发明还提供了一种孕酮受体靶向钌类配合物,其具有式(Ⅱ)所示结构:
其中,R表示的为
本发明还提供了所述孕酮受体靶向钌类配合物的制备方法,包括如下步骤:
室温避光条件下,将上述的方法所制得的式(Ⅰ)所示的孕酮受体靶向氮杂环卡宾中间体溶于配位反应溶剂中,加入Ag2O的催化活化,然后加入对花伞烃二氯化钌发生配位反应,过滤、重结晶,即得式(Ⅱ)所示的孕酮受体靶向钌类配合物。
进一步优选地,包括如下步骤:室温避光条件下,将上述的方法所制得的式(Ⅰ)所示的孕酮受体靶向氮杂环卡宾中间体溶于配位反应溶剂中,加入Ag2O的催化活化2-6h,然后加入对花伞烃二氯化钌反应2-6h,过滤、重结晶,即得式(Ⅱ)所示的孕酮受体靶向钌类配合物,其中,式(Ⅰ)所述的孕酮受体靶向氮杂环卡宾中间体、Ag2O和对花伞烃二氯化钌的摩尔比为1:(0.5-2):(0.5-2)。
进一步优选地,在重结晶步骤之后还包括半制备HPLC分离提纯步骤,其中,固定相为C18色谱柱,流动相A为含有0.1%TFA的去离子水,流动相B为含有0.1%TFA的乙腈,采用梯度洗脱收集产物,淋洗程序设定为:起始时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B。
进一步优选地,式(Ⅰ)所示的孕酮受体靶向氮杂环卡宾中间体、Ag2O和对花伞烃二氯化钌的摩尔比为1:(0.5-1):(0.5-1)。
进一步优选地,在重结晶过程中,采用正己烷和二氯甲烷、乙醚和二氯甲烷、正庚烷和二氯甲烷中的一种为重结晶试剂。
进一步优选地,在重结晶过程中,采用体积比为8:1-4:1的正己烷和二氯甲烷的混合溶剂为重结晶试剂。
本发明还提供了上述的方法所制备的配合物在制备靶向治疗乳腺癌药物中的应用。
进一步优选地,所述配合物抑制MCF-7的IC50值不大于抑制MDA-MB-231的IC50值的1/4,所述配合物在MCF-7细胞中的摄取值至少为在MDA-MB-231细胞中摄取值的10倍。
进一步优选地,所述配合物抑制MCF-7的IC50为不大于5μM,所述配合物对MCF-7细胞的摄取值为不小于0.2μg/106细胞。
进一步优选地,所述配合物在GSH浓度大于等于1mM能够断裂释放抗肿瘤药物,所述释放的抗肿瘤药物具有如下结构式:
其中,R表示的为
本发明还提供了一种药物制剂,以上述的方法所制备的配合物为活性成分,按照常规工艺,加入常规辅料制成临床上可接受的片剂、胶囊剂、散剂、合剂、丸剂、颗粒剂、糖浆剂、贴膏剂、栓剂、气雾剂、软膏剂或注射剂。
本发明还提供了上述的药物制剂在制备治疗靶向乳腺癌药物中的应用。
与现有技术相比,本发明的上述技术方案具有以下优点:
本发明所述的孕酮受体靶向钌类配合物,一方面,配合物中存在二硫键,其在高浓度的谷胱甘肽(GSH)中被还原,释放药物NHC-Ru,而在低浓度GSH中,二硫键并不能还原,不释放药物NHC-Ru,可以作为GSH特异性响应的药物;另一方面,细胞毒性及摄取实验显示,该配合物对孕酮受体阳性肿瘤细胞MCF-7(PR+)的IC50较低,摄取值较高,而对孕酮受体阴性肿瘤细胞MDA-MB-231(PR-)的IC50较高,摄取值较低,显示其对孕酮受体阳性肿瘤细胞具有较高的细胞毒性和靶向性;此外,动物实验也表明,该配合物对孕酮受体阳性肿瘤具有较高的靶向性及抗肿瘤活性,而且制备方法简单,易于提纯,该配合物可以作为靶向于PR受体的抗乳腺癌的药物。
附图说明
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中:
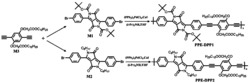
图1是本发明中式(Ⅱ)所示的孕酮受体靶向钌类配合物Pr-S-S-NHC-Ru的合成路线图;
图2是实施例1制得的式(Ⅰ)所示中间体Pr-S-S-NHC的质谱图;
图3是实施例1制得的式(Ⅱ)所示的孕酮受体靶向钌类配合物Pr-S-S-NHC-Ru的质谱图;
图4是实施例1制得的式(Ⅱ)所示的孕酮受体靶向钌类配合物Pr-S-S-NHC-Ru的
图5是实施例1制得的式(Ⅱ)所示的孕酮受体靶向钌类配合物Pr-S-S-NHC-Ru的
图6是对比例2制得的Pr-OH的质谱图;
图7是实验例1中式(Ⅱ)所示的孕酮受体靶向钌类配合物的GSH响应性试验的HPLC结果图;其中,A为钌类配合物GSH响应性原理图;B为不同GSH浓度下化合物的剪切情况的HPLC结果图;C为不同生物体系中化合物剪切情况的HPLC结果图;
图8是实验例1中式(Ⅱ)所示的孕酮受体靶向钌类配合物的细胞毒性和细胞摄取实验结果图;其中A为细胞毒性实验结果图,B为细胞摄取实验结果图。
图9是实验例1中式(Ⅱ)所示的孕酮受体靶向钌类配合物的小鼠体内抗肿瘤活性研究结果图;其中,A为给药0天和44天的小鼠;B为给药0-44天内肿瘤体积变化图;C为给药0-44天内小鼠体重变化图;D为给药0-44天后肿瘤小鼠的存活率结果图。
具体实施方式
下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1 式(Ⅱ)所示的孕酮受体靶向钌类配合物(Pr-S-S-NHC-Ru)的制备
本实施例化合物Pr-S-S-NHC-Ru的制备方法,按照图1所示的路线合成,具体包括以下步骤:
(1)NHC的制备:取4-羟甲基咪唑(1.34g,10mmol),K2CO3(4.14g,30mmol)和对叔丁基苄氯(1.82g,10mmol)在乙腈中加热回流8h,将反应液旋干,层析柱分离(CH2Cl2/CH3OH=10:1,v/v),得反应中间体a,其为淡黄色固体。然后将所得化合物a与对叔丁基苄氯(1.82g,10mmol)在乙腈中加热回流过夜,将乙腈旋干,层析柱分离(CH2Cl2/CH3OH=3:1,v/v),得白色固体NHC;
(2)N3-S-S-NHC的制备:将羰基二咪唑(1.62g,10mmol)和2-叠氮乙醇(0.87g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至2-羟乙基二硫化物(1.54g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(乙酸乙酯/正己烷=1:1,v/v),得油状液体b;将羰基二咪唑(1.62g,10mmol)和油状液体b(2.67g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至NHC(4.26g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(二氯甲烷/甲醇=10:1,v/v)),得油状液体N3-S-S-NHC;
(3)式(Ⅰ)所示中间体(Pr-S-S-NHC)的制备:取化合物N3-S-S-NHC(1.44g,2mmol)、炔孕酮(0.62g,2mmol)、CuSO4·5H2O(1.0g,4mmol)和抗坏血酸钠(0.79g,4mmol)溶于100mlH2O/MeOH(1:1,v/v))混合溶剂中,45℃下反应12h。然后柱层析分离纯化(8:1v/v二氯甲烷:MeOH),得Pr-S-S-NHC为淡黄色固体,产率55%;
鉴别:ESI-MS质谱分析结果为m/z=996.52([M]
(4)Pr-S-S-NHC-Ru的制备:室温避光条件下,将上述制得的化合物Pr-S-S-NHC(103mg,0.1mmol)、Ag2O(11.6mg,0.05mmol)溶于10ml二氯甲烷中,反应4h,然后加入对花伞烃二氯化钌(30.6mg,0.05mmol),继续反应4h。过滤,二氯甲烷/正己烷(v/v,1:4)混合溶剂中重结晶。
还包括半制备HPLC分离提纯步骤,取重结晶后得到的产品10mg,溶解于2ml甲醇中,使用Waters 2998 HPLC系统配备C18的HPLC分析柱(Phenomenex,5μm,250×19mm)梯度淋洗40分钟,流速3ml/min,波长254nm,其中流动相A为含0.1%TFA的去离子水,流动相B为含0.1%TFA的乙腈,梯度淋洗程序设定为:起始时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B,冻干即得式(Ⅱ)所示的化合物Pr-S-S-NHC-Ru,淡黄色色粉末,分子式:C65H87N5O8S2Cl2Ru,产率为26%,纯度大于98%。ESI-MS质谱分析结果为m/z=1266.55([M-Cl
实施例2 式(Ⅱ)所示的孕酮受体靶向钌类配合物(Pr-S-S-NHC-Ru)的制备
本实施例化合物Pr-S-S-NHC-Ru的制备方法,按照图1所示的路线合成,具体包括以下步骤:
(1)NHC的制备:取4-羟甲基咪唑(1.34g,10mmol),K2CO3(4.14g,30mmol)和对叔丁基苄氯(1.82g,10mmol)在THF中加热回流8h,将反应液旋干,层析柱分离(CH2Cl2/CH3OH=10:1,v/v),得反应中间体a,其为淡黄色固体。然后将所得化合物a与对叔丁基苄氯(1.82g,10mmol)在THF中加热回流过夜,将THF旋干,层析柱分离(CH2Cl2/CH3OH=3:1,v/v),得白色固体NHC;
(2)N3-S-S-NHC的制备:将羰基二咪唑(1.62g,10mmol)和2-叠氮乙醇(0.87g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至2-羟乙基二硫化物(1.54g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(乙酸乙酯/正己烷=1:1,v/v),得油状液体b;将羰基二咪唑(1.62g,10mmol)和油状液体b(2.67g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至NHC(4.26g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(二氯甲烷/甲醇=10:1,v/v)),得油状液体N3-S-S-NHC;
(3)式(Ⅰ)所示中间体(Pr-S-S-NHC)的制备:取化合物N3-S-S-NHC(1.44g,2mmol)、炔孕酮(1.24g,4mmol)、CuSO4·5H2O(1.0g,4mmol)和抗坏血酸钠(0.79g,4mmol)溶于100mlH2O/DMF(1:1,v/v))混合溶剂中,60℃下反应12h。然后柱层析分离纯化(8:1v/v二氯甲烷:MeOH),得Pr-S-S-NHC为淡黄色固体,产率54%;
鉴别:ESI-MS质谱分析结果为m/z=996.52([M
(4)Pr-S-S-NHC-Ru的制备:室温避光条件下,将上述制得的化合物Pr-S-S-NHC(103mg,0.1mmol)、Ag2O(23.2mg,0.1mmol)溶于10ml二氯甲烷中,反应6h,然后加入对花伞烃二氯化钌(61.2mg,0.1mmol),继续反应6h。过滤,二氯甲烷/正己烷(v/v,1:4)混合溶剂中重结晶。
还包括半制备HPLC分离提纯步骤,取重结晶后得到的产品10mg,溶解于2ml甲醇中,使用Waters 2998 HPLC系统配备C18的HPLC分析柱(Phenomenex,5μm,250×19mm)梯度淋洗40分钟,流速3ml/min,波长254nm,其中流动相A为含0.1%TFA的去离子水,流动相B为含0.1%TFA的乙腈,梯度淋洗程序设定为:起始时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B,冻干即得式(Ⅱ)所示的化合物Pr-S-S-NHC-Ru,淡黄色色粉末,分子式:C65H87N5O8S2Cl2Ru,产率为27%,纯度大于98%。ESI-MS质谱分析结果为m/z=1266.55([M-Cl
实施例3 式(Ⅱ)所示的孕酮受体靶向钌类配合物(Pr-S-S-NHC-Ru)的制备
本实施例化合物Pr-S-S-NHC-Ru的制备方法,按照图1所示的路线合成,具体包括以下步骤:
(1)NHC的制备:取4-羟甲基咪唑(1.34g,10mmol),K2CO3(4.14g,30mmol)和对叔丁基苄氯(1.82g,10mmol)在THF中加热回流8h,将反应液旋干,层析柱分离(CH2Cl2/CH3OH=10:1,v/v),得反应中间体a,其为淡黄色固体。然后将所得化合物a与对叔丁基苄氯(1.82g,10mmol)在THF中加热回流过夜,将THF旋干,层析柱分离(CH2Cl2/CH3OH=3:1,v/v),得白色固体NHC;
(2)N3-S-S-NHC的制备:将羰基二咪唑(1.62g,10mmol)和2-叠氮乙醇(0.87g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至2-羟乙基二硫化物(1.54g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(乙酸乙酯/正己烷=1:1,v/v),得油状液体b;将羰基二咪唑(1.62g,10mmol)和油状液体b(2.67g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至NHC(4.26g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(二氯甲烷/甲醇=10:1,v/v)),得油状液体N3-S-S-NHC;
(3)式(Ⅰ)所示中间体(Pr-S-S-NHC)的制备:取化合物N3-S-S-NHC(1.44g,2mmol)、炔孕酮(0.93g,3mmol)、CuSO4·5H2O(2.0g,8mmol)和抗坏血酸钠(1.58g,8mmol)溶于100mlH2O/乙腈(1:1,v/v))混合溶剂中,45℃下反应8h。然后柱层析分离纯化(8:1v/v二氯甲烷:MeOH),得Pr-S-S-NHC为淡黄色固体,产率53%;
鉴别:ESI-MS质谱分析结果为m/z=996.52([M]
(4)Pr-S-S-NHC-Ru的制备:室温避光条件下,将上述制得的化合物Pr-S-S-NHC(103mg,0.1mmol)、Ag2O(46.4mg,0.2mmol)溶于10ml二氯甲烷中,反应2h,然后加入对花伞烃二氯化钌(122.4mg,0.2mmol),继续反应2h。过滤,二氯甲烷/乙醚(v/v,1:4)混合溶剂中重结晶。
还包括半制备HPLC分离提纯步骤,取重结晶后得到的产品10mg,溶解于2ml甲醇中,使用Waters 2998 HPLC系统配备C18的HPLC分析柱(Phenomenex,5μm,250×19mm)梯度淋洗40分钟,流速3ml/min,波长254nm,其中流动相A为含0.1%TFA的去离子水,流动相B为含0.1%TFA的乙腈,梯度淋洗程序设定为:起始时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B,冻干即得式(Ⅱ)所示的化合物Pr-S-S-NHC-Ru,淡黄色色粉末,分子式:C65H87N5O8S2Cl2Ru,产率为23%,纯度大于98%。ESI-MS质谱分析结果为m/z=1266.55([M-Cl
实施例4 式(Ⅱ)所示的孕酮受体靶向钌类配合物(Pr-S-S-NHC-Ru)的制备
本实施例化合物Pr-S-S-NHC-Ru的制备方法,按照图1所示的路线合成,具体包括以下步骤:
(1)NHC的制备:取4-羟甲基咪唑(1.34g,10mmol),K2CO3(4.14g,30mmol)和对叔丁基苄氯(1.82g,10mmol)在THF中加热回流8h,将反应液旋干,层析柱分离(CH2Cl2/CH3OH=10:1,v/v),得反应中间体a,其为淡黄色固体。然后将所得化合物a与对叔丁基苄氯(1.82g,10mmol)在THF中加热回流过夜,将THF旋干,层析柱分离(CH2Cl2/CH3OH=3:1,v/v),得白色固体NHC;
(2)N3-S-S-NHC的制备:将羰基二咪唑(1.62g,10mmol)和2-叠氮乙醇(0.87g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至2-羟乙基二硫化物(1.54g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(乙酸乙酯/正己烷=1:1,v/v),得油状液体b;将羰基二咪唑(1.62g,10mmol)和油状液体b(2.67g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至NHC(4.26g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(二氯甲烷/甲醇=10:1,v/v)),得油状液体N3-S-S-NHC;
(3)式(Ⅰ)所示中间体(Pr-S-S-NHC)的制备:取化合物N3-S-S-NHC(1.44g,2mmol)、炔孕酮(0.74g,2.4mmol)、六氟磷酸四乙腈铜(1.12g,3mmol)溶于100mlH2O/MeOH(1:1,v/v))混合溶剂中,37℃下反应20h。然后柱层析分离纯化(8:1v/v二氯甲烷:MeOH),得Pr-S-S-NHC为淡黄色固体,产率58%;
鉴别:ESI-MS质谱分析结果为m/z=996.52([M]
(4)Pr-S-S-NHC-Ru的制备:室温避光条件下,将上述制得的化合物Pr-S-S-NHC(103mg,0.1mmol)、Ag2O(23.2mg,0.1mmol)溶于10ml二氯甲烷中,反应6h,然后加入对花伞烃二氯化钌(61.2mg,0.1mmol),继续反应4h。过滤,二氯甲烷/正己烷(v/v,1:4)混合溶剂中重结晶。
还包括半制备HPLC分离提纯步骤,取重结晶后得到的产品10mg,溶解于2ml甲醇中,使用Waters 2998 HPLC系统配备C18的HPLC分析柱(Phenomenex,5μm,250×19mm)梯度淋洗40分钟,流速3ml/min,波长254nm,其中流动相A为含0.1%TFA的去离子水,流动相B为含0.1%TFA的乙腈,梯度淋洗程序设定为:起始时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B,冻干即得式(Ⅱ)所示的化合物Pr-S-S-NHC-Ru,淡黄色色粉末,分子式:C65H87N5O8S2Cl2Ru,产率为27%,纯度大于98%。ESI-MS质谱分析结果为m/z=1266.55([M-Cl
实施例5 式(Ⅱ)所示的孕酮受体靶向钌类配合物(Pr-S-S-NHC-Ru)的制备
本实施例化合物Pr-S-S-NHC-Ru的制备方法,按照图1所示的路线合成,具体包括以下步骤:
(1)NHC的制备:取4-羟甲基咪唑(1.34g,10mmol),K2CO3(4.14g,30mmol)和对叔丁基苄氯(1.82g,10mmol)在THF中加热回流8h,将反应液旋干,层析柱分离(CH2Cl2/CH3OH=10:1,v/v),得反应中间体a,其为淡黄色固体。然后将所得化合物a与对叔丁基苄氯(1.82g,10mmol)在THF中加热回流过夜,将THF旋干,层析柱分离(CH2Cl2/CH3OH=3:1,v/v),得白色固体NHC;
(2)N3-S-S-NHC的制备:将羰基二咪唑(1.62g,10mmol)和2-叠氮乙醇(0.87g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至2-羟乙基二硫化物(1.54g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(乙酸乙酯/正己烷=1:1,v/v),得油状液体b;将羰基二咪唑(1.62g,10mmol)和油状液体b(2.67g,10mmol)溶于50ml二氯甲烷中,室温反应30min。然后将其慢慢滴加至NHC(4.26g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(二氯甲烷/甲醇=10:1,v/v)),得油状液体N3-S-S-NHC;
(3)式(Ⅰ)所示中间体(Pr-S-S-NHC)的制备:取化合物N3-S-S-NHC(1.44g,2mmol)、炔孕酮(0.62g,2mmol)、氧化亚铜(1.0g,4mmol)和醋酸(0.79g,4mmol)溶于100mlH2O/乙醇(1:1,v/v))混合溶剂中,45℃下反应12h。然后柱层析分离纯化(8:1v/v二氯甲烷:MeOH),得Pr-S-S-NHC为淡黄色固体,产率55%;
鉴别:ESI-MS质谱分析结果为m/z=996.52([M]
(4)Pr-S-S-NHC-Ru的制备:室温避光条件下,将上述制得的化合物Pr-S-S-NHC(103mg,0.1mmol)、Ag2O(11.6mg,0.05mmol)溶于10ml二氯甲烷中,反应2h,然后加入对花伞烃二氯化钌(30.6mg,0.05mmol),继续反应2h。过滤,二氯甲烷/正庚烷(v/v,1:8)混合溶剂中重结晶。
还包括半制备HPLC分离提纯步骤,取重结晶后得到的产品10mg,溶解于2ml甲醇中,使用Waters 2998 HPLC系统配备C18的HPLC分析柱(Phenomenex,5μm,250×19mm)梯度淋洗40分钟,流速3ml/min,波长254nm,其中流动相A为含0.1%TFA的去离子水,流动相B为含0.1%TFA的乙腈,梯度淋洗程序设定为:起始时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B,冻干即得式(Ⅱ)所示的化合物Pr-S-S-NHC-Ru,淡黄色色粉末,分子式:C65H87N5O8S2Cl2Ru,产率为24%,纯度大于98%。ESI-MS质谱分析结果为m/z=1266.55([M-Cl
对比例1 Pr-S-S-NHC-Ru的制备
本对比例1的Pr-S-S-NHC-Ru的拟合成路线及具体步骤如下:
其中,R表示的为
NHC的制备:取4-羟甲基咪唑(1.34g,10mmol),K2CO3(4.14g,30mmol)和对叔丁基苄氯(1.82g,10mmol)在乙腈中加热回流8h,将反应液旋干,层析柱分离(CH2Cl2/CH3OH=10:1,v/v),得反应中间体a,其为淡黄色固体。然后将所得化合物a与对叔丁基苄氯(1.82g,10mmol)在乙腈中加热回流过夜,将乙腈旋干,层析柱分离(CH2Cl2/CH3OH=3:1,v/v),得白色固体NHC。
化合物3的制备:将羰基二咪唑(1.62g,10mmol)和化合物1(4.26g,10mmol)溶于50ml二氯甲烷中,室温反应30min,得中间体化合物2。不经分离,然后将其慢慢滴加至2-羟乙基二硫化物(1.54g,10mmol)的二氯甲烷溶液中,室温反应12h。反应结束后,将溶剂旋干,层析柱分离(二氯甲烷/甲醇=10:1,v/v)),得油状液体化合物3。
叠氮乙醇的制备:2-溴乙醇(1.25g,10mmol)和叠氮化钠(1.95g,30mmol)溶于50mL水中,90℃下反应过夜。冷至室温后,二氯甲烷萃取,有机相用无水硫酸钠干燥,旋干,得叠氮乙醇为无色油状液体。产率87%
化合物4的制备:取化合物叠氮乙醇(0.17g,2mmol)、炔孕酮(0.62g,2mmol)、CuSO4·5H2O(1.0g,4mmol)和抗坏血酸钠(0.79g,4mmol)溶于100mlH2O/MeOH(1:1,v/v)混合溶剂中,45℃下反应12h。然后柱层析分离纯化(8:1v/v二氯甲烷:MeOH),得化合物4为白色固体,产率65%;
化合物5的制备:将羰基二咪唑(1.62g,10mmol)和化合物4(3.99g,10mmol)溶于50ml二氯甲烷中,室温反应30min,得化合物5为白色固体,产率83%。
化合物5与化合物3混合后无法完成后续反应,导致化合物6无法合成,无法进行后续步骤合成。
对比例2 NHC-Ru的制备
本对比例2 NHC-Ru的合成路线及具体步骤如下:
(1)NHC的制备:取4-羟甲基咪唑(1.34g,10mmol),K2CO3(4.14g,30mmol)和对叔丁基苄氯(1.82g,10mmol)在乙腈中加热回流8h,将反应液旋干。层析柱分离(CH2Cl2/CH3OH=10:1,v/v),得中间体a为淡黄色固体。然后将所得化合物a与对叔丁基苄氯(1.82g,10mmol)在乙腈中加热回流过夜,将乙腈旋干,层析柱分离(CH2Cl2/CH3OH=3:1,v/v),得白色固体NHC;
(2)NHC-Ru的制备:室温避光条件下,将上述制得的化合物NHC(42.6mg,0.1mmol)、Ag2O(11.6mg,0.05mmol)溶于10ml二氯甲烷中,反应4h,然后加入对花伞烃二氯化钌(30.6mg,0.05mmol),继续反应4h。过滤,1:4v/v二氯甲烷:正己烷混合溶剂重结晶后得到化合物NHC-Ru;
还包括半制备HPLC分离提纯步骤,取重结晶后得到的化合物NHC-Ru10mg,溶解于2ml甲醇中,使用Waters 2998 HPLC系统配备C18HPLC分析柱(Phenomenex,5μm,250×19mm)梯度淋洗40分钟,流速3ml/min,波长254nm,其中流动相A为含0.1%TFA的去离子水,流动相B为含0.1%TFA的乙腈。梯度淋洗程序设定为:起始时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B,冻干即得化合物NHC-Ru。
ESI-MS质谱分析结果为m/z=666.5([M-Cl]
实验例1 式(Ⅱ)所示的孕酮受体靶向钌类配合物的GSH响应性试验
1、实验目的
检测式(Ⅱ)所示的化合物Pr-S-S-NHC-Ru在不同GSH浓度下药物NHC-Ru的释放情况。
2、实验方法
2.1实验仪器和试剂
Waters515泵,紫外检测器(Waters 2487)。
色谱柱:PhenomenexLunaC-18(5μm,250×4.6mm)分析柱。
流动相:流动相A为含0.1%三氟乙酸的水溶液,流动相B为含0.1%三氟乙酸的乙腈溶液。
梯度淋洗程序设定为:起始时,80%A和20%B,10min时,40%A和60%B;35min时,10%A和90%B;40min时,80%A和20%B;流速1.0ml/min,检测波长254nm。
2.2实验方法
(1)Pr-OH的制备
取化合物2-叠氮乙醇(0.174g,2mmol)、炔孕酮(0.62g,2mmol)、CuSO4·5H2O(1.0g,4mmol)和抗坏血酸钠(0.79g,4mmol)溶于100mlH2O/MeOH(1:1,v/v)混合溶剂中,45℃下反应12h。然后柱层析分离纯化(8:1v/v二氯甲烷:MeOH),得Pr-OH固体。
ESI-MS质谱分析结果为m/z=400.92([M+H]
(3)不同GSH浓度下化合物的剪切情况:
平行条件下精密量取5组化合物Pr-S-S-NHC-Ru,每组均含有5μl,0.1mM的Pr-S-S-NHC-Ru,作为试验组,分别加入到100μlPBS(1×,pH7.4)中,然后加入不同浓度GSH(0,10μM,100μM,1mM,5mM);同时取空白对照组Pr-OH(5μl,0.1mM)和阴性对照组NHC-Ru(5μl,0.1mM)分别加入到100μlPBS(1×,pH7.4)中,将5组试验组和2组对照组分别置于37℃下孵育1h,然后各取反应液20μl,注入液相色谱仪,测定剪切情况。
(4)不同生物体系中化合物的剪切情况:
平行条件下精密量取多组Pr-S-S-NHC-Ru,每组均含有0.1mM的Pr-S-S-NHC-Ru(5μl),作为试验组,分别加入到100μlPBS(1×,pH7.4)中,各组不加试剂或者分别加入5mMCys(半胱氨酸)、Gly(甘氨酸)、Ala(丙氨酸)、Lys(赖氨酸)、Ser(丝氨酸)、Glu(谷氨酸)、Na
3、实验结果
如图1所示,在低浓度GSH条件下(10-100μM),化合物Pr-S-S-NHC-Ru的HPLC并未发生变化,这说明二硫键并未被还原;而当GSH浓度为1mM时,出现了两个新的峰,分别对应剪切之后的化合物Pr-OH及NHC-Ru。当GSH浓度为5mM时,化合物Pr-S-S-NHC-Ru的峰消失,二硫键全部被还原(如图3所示)。
如图3所示,不同生物体系对化合物Pr-S-S-NHC-Ru的剪切情况,其中,由于半胱氨酸(Cys)中同样含有巯基,因此其在高浓度下可以剪切二硫键;但Cys在细胞中含量较低,不会影响药物的释放。而在其它生物体系中,化合物Pr-S-S-NHC-Ru均不能被还原。这说明该化合物可以作为GSH特异性响应的药物。
实验例2 式(Ⅱ)所示的孕酮受体靶向钌类配合物的细胞毒性实验
1、实验目的
研究化合物Pr-S-S-NHC-Ru和NHC-Ru对MCF-7(PR+)及MDA-MB-231(PR-)的细胞毒性。
2、实验方法
采用MTT法测定细胞存活率,根据配合物对细胞生长的半抑制浓度(IC50)值,衡量配合物的抗癌活性。于96孔板中接种处于对数生长期的细胞,细胞接种浓度为7×10
3、实验结果
结果见图7所示,对孕酮受体阳性肿瘤细胞MCF-7(PR+),化合物Pr-S-S-NHC-Ru表现出更好的活性。尤其在6.125μM时,两者表现出较大差异。而对孕酮受体阴性肿瘤细胞MDA-MB-231(PR-),两者活性相当。化合物Pr-S-S-NHC-Ru和NHC-Ru对MCF-7(PR+)的IC50值分别为4.48±0.17和10.54±0.34μM,及对MDA-MB-231(PR-)的IC50值分别为20.71±0.92和14.18±1.01μM。化合物Pr-S-S-NHC-Ru对MCF-7和MDA-MB-231细胞表现出明显的活性差异,这主要是由于其靶向基团孕酮与肿瘤细胞中孕酮受体的特异性结合造成的。
实验例3 式(Ⅱ)所示的孕酮受体靶向钌类配合物的细胞摄取实验
1、实验目的
研究MCF-7(PR+)及MDA-MB-231(PR-)对化合物Pr-S-S-NHC-Ru和NHC-Ru的细胞摄取情况。
2、实验方法
将细胞接种于10cm的培养皿中,接种数目为10
3、实验结果
结果见图7所示,在MCF-7细胞中,化合物Pr-S-S-NHC-Ru的摄取值要高于化合物NHC-Ru,分别为0.21±0.01和0.053±0.008μg/10
实验例4 式(Ⅱ)所示的孕酮受体靶向钌类配合物的体内活性研究
1、实验目的
研究Pr-S-S-NHC-Ru的体内抗肿瘤活性。
2、实验方法
4-5周大小的雌性BALB/c裸鼠,喂养含0.3mg/ml雌激素的水一周,然后在右前肢接种0.1ml含2×10
3、实验结果
如图9所示,44天后,对照组小鼠肿瘤体积明显增加,而Pr-S-S-NHC-Ru治疗后,小鼠肿瘤体积从144±25mm
综上,本发明所述的孕酮受体靶向钌类配合物对孕酮受体阳性肿瘤细胞MCF-7(PR+)的IC50较低,摄取值较高,而对孕酮受体阴性肿瘤细胞MDA-MB-231(PR-)的IC50较高,摄取值较低,显示其对孕酮受体阳性肿瘤细胞具有较高的靶向性和较高的抗肿瘤作用;此外,动物实验也表明,该配合物对孕酮受体阳性肿瘤具有较高的靶向性及抗肿瘤活性,降低毒副作用,提高小鼠的生存率,有利于制备得到靶向于PR受体的抗乳腺癌药物在临床上推广应用。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
一种孕酮受体靶向氮杂环卡宾中间体和钌类配合物及其制备方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0