
-一枝蒿碱G的全合成及对映异构体的拆分方法")
IPC分类号 : C07D213/61,C07D221/04,C07B57/00,C07D213/84



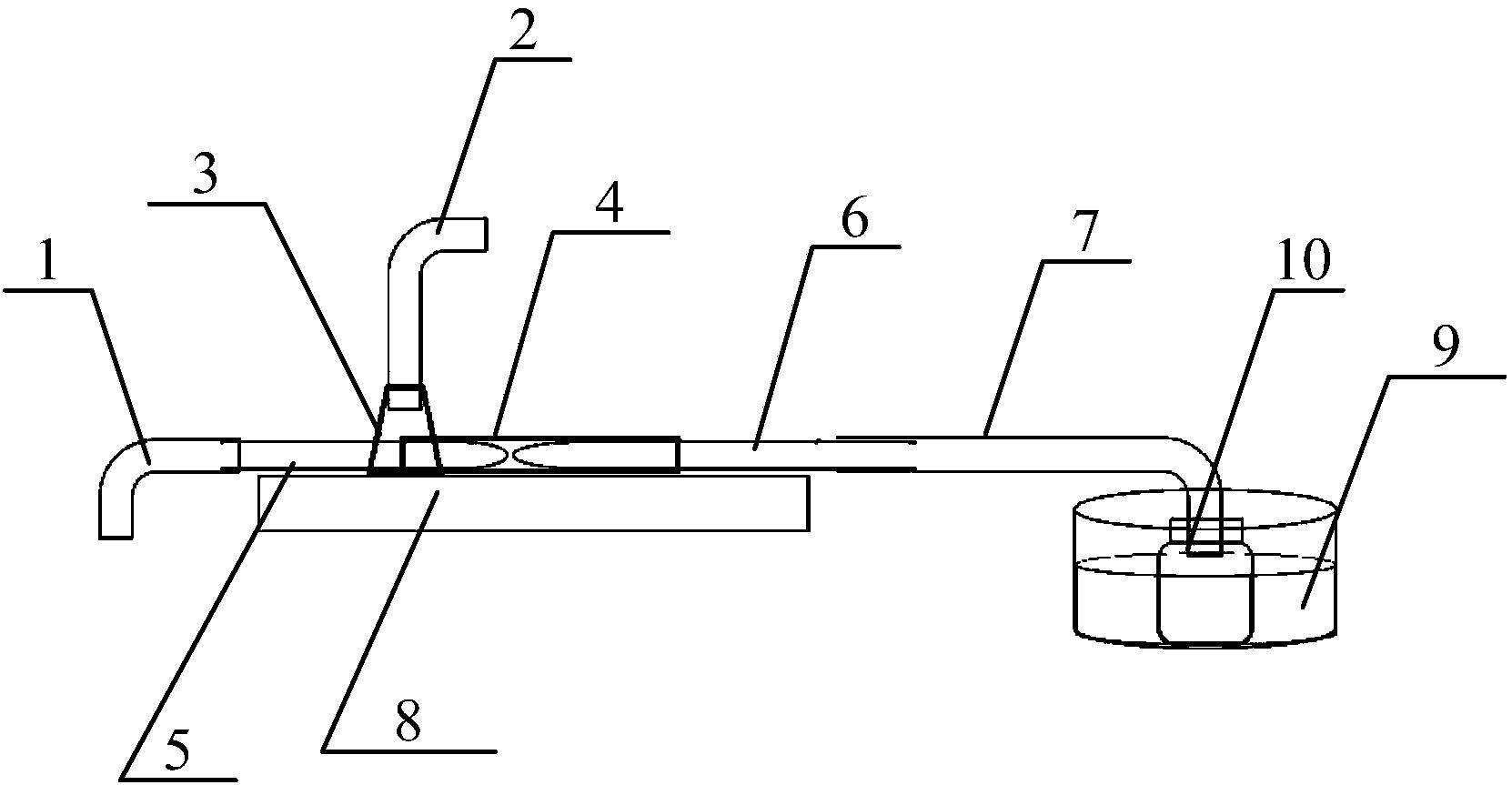


专利摘要
本发明涉及一种天然产物(±)‑一枝蒿碱G的全合成及对映异构体的拆分方法,该方法以2‑甲基‑5‑溴吡啶(1)为原料,通过间氯过氧苯甲酸氧化得到氮氧化产物化合物2,经Reissert‑Henze反应氰基取代得到化合物3,将化合物3和丙二酸单乙酯钾盐通过Blaise反应得到β‑酮酯4,在乙醇钠条件下烷基化得到化合物5,经偶联反应得到化合物6,将化合物6发生分子内烯烃复分解反应得到关键中间体化合物7,用硼氢化钠还原得到化合物8,在吡啶/MsCl条件下得到化合物9,化合物9在氢氧化锂作用下水解,再用碘甲烷甲酯化得到化合物10,再通过钯碳催化氢化得到一对非对映异构体11和12,再经半制备型高效液相色谱仪进行拆分,得到天然产物一枝蒿碱G以及三个立体异构体化合物13、化合物14和化合物15。
权利要求
1.一种天然产物(±)-一枝蒿碱G的全合成及对映异构体的拆分方法,其特征在于,所述的合成方法是由廉价易得的工业原料2-甲基-5-溴吡啶经过有机合成反应,包括氰基取代、脱羧Blaise反应、Suzuki偶联和烯烃复分解反应,最后得到(±)-一枝蒿碱G及其非对映异构体,再用半制备型高效液相色谱仪,进行对应异构体的拆分,其结构式为:
其中:
化合物4为:3-(3-溴-6-甲基吡啶)-3-羰基丙酸乙酯;
化合物5为:2-(3-溴-6-甲基吡啶-2-甲酰基)-4-烯戊酸乙酯;
化合物6为:2-(3-异丙烯基-6-甲基吡啶-2-甲酰基)-4-戊烯酸乙酯;
化合物7为:2,5-二甲基-5-烯-9-氧代-环庚烷并[b]吡啶-8-甲酸乙酯;
化合物8为:2,5-二甲基-5-烯-9-羟基-环庚烷并[b]吡啶-8-甲酸乙酯;
化合物9为:2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸乙酯;
化合物10为:2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸甲酯;
化合物11为:反-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基;
化合物12为:顺-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基;
化合物13为:(5S,8R)-一枝蒿碱G;
化合物14为:(5R,8S)-一枝蒿碱G;
化合物15为:(5R,8R)-一枝蒿碱G;
具体操作按下列步骤进行:
a、将化合物2-甲基-5-溴吡啶(1)溶于二氯甲烷溶液中,冰浴下分批加入间氯过氧苯甲酸后,自然升温至室温,搅拌过夜反应,反应结束后将反应液倒入到饱和的亚硫酸钠溶液中淬灭,搅拌1小时之后用二氯甲烷萃取3次,合并有机相,用无水硫酸镁干燥,除去有机溶剂后得到氮氧化产物化合物2为5-溴-2-甲基-N-氧吡啶;
b、在氮气保护下,将化合物2为5-溴-2-甲基-N-氧吡啶溶解于乙腈溶液中,依次加入三甲基氰硅烷和三乙胺,回流12小时后,除去有机溶剂,纯化,得到化合物3为3-溴-6-甲基-2-氰基吡啶;
c、真空状态下,将含结晶水的氯化锌加热至熔融除水,在氮气保护下,依次加入1,2-二氯乙烷、化合物3为3-溴-6-甲基-2-氰基吡啶、丙二酸单乙酯钾盐和二异丙基乙胺,搅拌回流12小时,待体系冷却至室温后加入6N盐酸,再次回流1小时,分出有机相,水相用二氯甲烷萃取3次,合并有机相后用无水硫酸镁干燥,除去有机溶剂,纯化,得到化合物4为:3-(3-溴-6-甲基吡啶)-3-羰基丙酸乙酯;
d、将化合物4为3-(3-溴-6-甲基吡啶)-3-羰基丙酸乙酯溶解在无水乙醇中,冰浴中加入乙醇钠,搅拌的同时加入3-溴丙烯,室温过夜反应,除去有机溶剂,柱层析纯化,得到化合物5为:2-(3-溴-6-甲基吡啶-2-甲酰基)-4-烯戊酸乙酯;
e、将化合物5为2-(3-溴-6-甲基吡啶-2-甲酰基)-4-烯戊酸乙酯溶解于二氧六环和水的混合溶液中,加入四三苯基膦钯催化剂、碳酸钠、异丙烯基硼酸频哪醇酯,搅拌加热回流3小时,待体系冷却至室温后除去溶剂,柱层析纯化,得到化合物6为:2-(3-异丙烯基-6-甲基吡啶-2-甲酰基)-4-戊烯酸乙酯;
f、在氮气保护下,将化合物6为2-(3-异丙烯基-6-甲基吡啶-2-甲酰基)-4-戊烯酸乙酯溶解于二氯甲烷溶液中,加入Grubbs二代催化剂,加热回流反应12小时,待体系冷却至室温,除去有机溶剂,柱层析,得到化合物7为:2,5-二甲基-5-烯-9-氧代-环庚烷并[b]吡啶-8-甲酸乙酯;
g、在冰浴下,将化合物7为2,5-二甲基-5-烯-9-氧代-环庚烷并[b]吡啶-8-甲酸乙酯溶解于无水甲醇中,加入硼氢化钠,自然升温至室温,反应3小时后,加水淬灭反应,除去溶剂,柱层析,纯化得到化合物8为:2,5-二甲基-5-烯-9-羟基-环庚烷并[b]吡啶-8-甲酸乙酯;
h、将化合物8为2,5-二甲基-5-烯-9-羟基-环庚烷并[b]吡啶-8-甲酸乙酯溶于吡啶中,加热至温度60℃,再加入MsCl,反应3小时后,冷却至室温,加水淬灭反应,再用二氯甲烷萃取分出有机相,有机相用无水硫酸镁干燥,柱层析纯化,得到化合物9为:2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸乙酯;
i、将化合物9为2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸乙酯溶于四氢呋喃和水的混合溶液中,加入氢氧化锂,室温搅拌过夜反应,除去溶剂,所得的粗产物溶于无水甲醇中,冰浴下加入碘甲烷和1,8-二氮杂双环[5.4.0]十一碳-7-烯,室温搅拌过夜反应,后除去溶剂,柱层析,得到化合物10为:2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸甲酯;
j、将化合物10为2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸甲酯溶于无水甲醇中,冰浴下加入钯碳催化剂,通入氢气,室温搅拌过夜反应,除去溶剂,用薄层色谱层析方法分离纯化,得到化合物11为:反-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基和化合物12为:顺-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基;
k、将化合物11为反-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基和化合物12为顺-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基分别用半制备型高效液相色谱法拆分,得到化合物13(5S,8R)-一枝蒿碱G;化合物14(5R,8S)-一枝蒿碱G;化合物15(5R,8R)-一枝蒿碱G;一枝蒿碱G。
说明书
技术领域
本发明涉及有机合成技术和分离技术领域,尤其涉及一种天然产物一枝蒿碱G(Rupestine G)的全合成方法及对映异构体拆分方法。
背景技术
2008年,中国科学院新疆理化技术研究所阿吉艾克拜尔·艾萨研究员课题组首次从新疆一枝蒿中分离到了五个结构新颖的愈创吡啶型倍半萜类生物碱。2012年,该课题组的贺飞等从新疆一枝蒿中又分离得到八个该类型的化合物。从结构上看,该类型的化合物有一个吡啶环,一个饱和的七元环,具有两个手性中心。据文献报道,愈创吡啶类倍半萜生物碱具有肝癌细胞毒性,而新疆一枝蒿具有抗病毒,护肝等功效。这引起了众多合成化学家,生物家以及病理学家的极大兴趣。但是截止目前,除了该课题组外,未见其他个人或单位报道该化合物的全合成研究。本发明为进一步深入研究该类化合物的活性研究提供了有力保障。
参考文献
1.Su,Z.;Wu,H.;Yang,Y.;Aisa,H.A.;Slukhan,U.;Aripova,S.,Preparative isolation of guaipyridine sesquiterpene alkaloid from Artemisia rupestris L.flowers using high-speed counter-current chromatography.J.Sep.Sci.2008,31(12),2161-6.
2.Su,Z.;Wu,H.;Yang,Y.;Aisa,H.A.;Slukhan,U.;Aripova,S.,New Guaipyridine Sesquiterpene Alkaloids from Artemisia rupestris L.Helv.Chim.Acta 2010,93,33-38.
3.He,F.;Nugroho,A.E.;Wong,C.P.;Hirasawa,Y.;Shirota,O.;Morita,H.;Aisa,H.A.,Rupestines F—M,New Guaipyridine Sesquiterpene Alkaloids from Artemisia rupestris.Chem.Pharm.Bull.2012,60(2),213-218.
4.Tian-Jye Hsieh;Fang-Rong Chang;Yi-Chen Chia;Chung-Yi Chen;Hui-Fen Chiu;Wu,Y.-C.,Cytotoxic Constituents of the Fruits of Cananga odorata.J.Nat.Prod.2001,64,616-619.
5.Koyama,J.;Okatani,T.;Tagahara,K.;Suzuta,V.,Synthesis of guaipyridine,epiguaipyridine and related compounds.Heterocycles 1987,26(4),926-927.
6.Craig,D.;Henry,G.D.,Total Synthesis of the Cytotoxic Guaipyridine Sesquiterpene Alkaloid(+)-Cananodine.Eur.J.Org.Chem.2006,16,3558-3561.
本发明在国内外有关专利、文献的综合分析的基础上,以2-甲基-5-溴吡啶为原料,经过一系列的有机合成反应成功构建愈创吡啶类二萜生物碱的骨架,得到一种天然产物Rupestine G的同时分离拆分得到其三个异构体。
发明内容
本发明的目的在于,提供一种一种天然产物(±)-一枝蒿碱G的全合成及对映异构体的拆分方法,并且对其绝对构型进行确定。该方法以2-甲基-5-溴吡啶为原料,通过间氯过氧苯甲酸氧化得到氮氧化产物化合物2,将化合物2经Reissert-Henze反应氰基取代得到化合物3,将化合物3和丙二酸单乙酯钾盐通过脱羧Blaise反应得到β-酮酯4,将化合物4在乙醇钠条件下烷基化得到化合物5,进一步Suzuki偶联反应得到化合物6,将化合物6发生分子内烯烃复分解反应得到关键中间体化合物7,化合物7用硼氢化钠还原得到化合物8,最后化合物8在吡啶/MsCl条件下消除得到化合物9,将化合物9溶于四氢呋喃和水(v/v=1:1)中用氢氧化锂水解,后与碘甲烷在1,8-二氮杂双环[5.4.0]十一碳-7-烯存在下烷基化得到化合物10,将化合物10通过钯碳催化氢化得到一对非对映异构体11和12(m16/m17=1:1),外消旋体化合物11和12用SHIMADZU LC20A半制备型高效液相用CHIRALPAK ID(Lot No.ID00CE-QI011)拆分,分别得到化合物13和化合物14及一枝蒿碱G(Rupestine G)和化合物15。
本发明所述的一种天然产物(±)-一枝蒿碱G的全合成及对映异构体的拆分方法,其特征在于,所述的合成方法是由廉价易得的工业原料2-甲基-5-溴吡啶经过有机合成反应,包括氰基取代、脱羧Blaise反应、Suzuki偶联和烯烃复分解反应,最后得到(±)-一枝蒿碱G及其非对映异构体,再用半制备型高效液相色谱仪,进行对应异构体的拆分,其结构式为:
其中:
化合物4为:3-(3-溴-6-甲基吡啶)-3-羰基丙酸乙酯;
化合物5为:2-(3-溴-6-甲基吡啶-2-甲酰基)-4-烯戊酸乙酯;
化合物6为:2-(3-异丙烯基-6-甲基吡啶-2-甲酰基)-4-戊烯酸乙酯;
化合物7为:2,5-二甲基-5-烯-9-氧代-环庚烷并[b]吡啶-8-甲酸乙酯;
化合物8为:2,5-二甲基-5-烯-9-羟基-环庚烷并[b]吡啶-8-甲酸乙酯;
化合物9为:2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸乙酯;
化合物10为:2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸甲酯;
化合物11为:反-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基;
化合物12为:顺-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基;
化合物13为:(5S,8R)-一枝蒿碱G;
化合物14为:(5R,8S)-一枝蒿碱G;
化合物15为:(5R,8R)-一枝蒿碱G;
具体操作按下列步骤进行:
a、将化合物1为2-甲基-5-溴吡啶溶于二氯甲烷溶液中,冰浴下分批加入间氯过氧苯甲酸后,自然升温至室温,搅拌过夜反应,反应结束后将反应液倒入到饱和的亚硫酸钠溶液中淬灭,搅拌1小时之后用二氯甲烷萃取3次,合并有机相,用无水硫酸镁干燥,除去有机溶剂后得到氮氧化产物化合物2为5-溴-2-甲基-N-氧吡啶;
b、在氮气保护下,将化合物2为5-溴-2-甲基-N-氧吡啶溶解于乙腈溶液中,依次加入三甲基氰硅烷和三乙胺,回流12小时后,除去有机溶剂,纯化,得到化合物3为3-溴-6-甲基-2-氰基吡啶;
c、真空状态下,将含结晶水的氯化锌加热至熔融除水,在氮气保护下,依次加入1,2-二氯乙烷、化合物3为3-溴-6-甲基-2-氰基吡啶、丙二酸单乙酯钾盐和二异丙基乙胺,搅拌回流12小时,待体系冷却至室温后加入6N盐酸,再次回流1小时,分出有机相,水相用二氯甲烷萃取3次,合并有机相后用无水硫酸镁干燥,除去有机溶剂,纯化,得到化合物4为:3-(3-溴-6-甲基吡啶)-3-羰基丙酸乙酯;
d、将化合物4为3-(3-溴-6-甲基吡啶)-3-羰基丙酸乙酯溶解在无水乙醇中,冰浴中加入乙醇钠,搅拌的同时加入3-溴丙烯,室温过夜反应,除去有机溶剂,柱层析纯化,得到化合物5为:2-(3-溴-6-甲基吡啶-2-甲酰基)-4-烯戊酸乙酯;
e、将化合物5为2-(3-溴-6-甲基吡啶-2-甲酰基)-4-烯戊酸乙酯溶解于二氧六环和水的混合溶液中,加入四三苯基膦钯催化剂、碳酸钠、异丙烯基硼酸频哪醇酯,搅拌加热回流3小时,待体系冷却至室温后除去溶剂,柱层析纯化,得到化合物6为:2-(3-异丙烯基-6-甲基吡啶-2-甲酰基)-4-戊烯酸乙酯;
f、在氮气保护下,将化合物6为2-(3-异丙烯基-6-甲基吡啶-2-甲酰基)-4-戊烯酸乙酯溶解于二氯甲烷溶液中,加入Grubbs二代催化剂,加热回流反应12小时,待体系冷却至室温,除去有机溶剂,柱层析,得到化合物7为:2,5-二甲基-5-烯-9-氧代-环庚烷并[b]吡啶-8-甲酸乙酯;
g、在冰浴下,将化合物7为2,5-二甲基-5-烯-9-氧代-环庚烷并[b]吡啶-8-甲酸乙酯溶解于无水甲醇中,加入硼氢化钠,自然升温至室温,反应3小时后,加水淬灭反应,除去溶剂,柱层析,纯化得到化合物8为:2,5-二甲基-5-烯-9-羟基-环庚烷并[b]吡啶-8-甲酸乙酯;
h、将化合物8为2,5-二甲基-5-烯-9-羟基-环庚烷并[b]吡啶-8-甲酸乙酯溶于吡啶中,加热至温度60℃,再加入MsCl,反应3小时后,冷却至室温,加水淬灭反应,再用二氯甲烷萃取分出有机相,有机相用无水硫酸镁干燥,柱层析纯化,得到化合物9为:2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸乙酯;
i、将化合物9为2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸乙酯溶于四氢呋喃和水的混合溶液中,加入氢氧化锂,室温搅拌过夜反应,除去溶剂,所得的粗产物溶于无水甲醇中,冰浴下加入碘甲烷和1,8-二氮杂双环[5.4.0]十一碳-7-烯,室温搅拌过夜反应,后除去溶剂,柱层析,得到化合物10为:2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸甲酯;
j、将化合物10为2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸甲酯溶于无水甲醇中,冰浴下加入钯碳催化剂,通入氢气,室温搅拌过夜反应,除去溶剂,用薄层色谱层析方法分离纯化,得到化合物11为:反-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基和化合物12为:顺-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基;
k、将化合物11为反-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基和化合物12为顺-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基分别用半制备型高效液相色谱法拆分,得到化合物13(5S,8R)-一枝蒿碱G;化合物14为:(5R,8S)-一枝蒿碱G;化合物15为:(5R,8R)-一枝蒿碱G;一枝蒿碱G。
本发明所述的一种天然产物(±)-一枝蒿碱G的全合成及对映异构体的拆分方法,该方法的合成路线如下所示:
附图说明
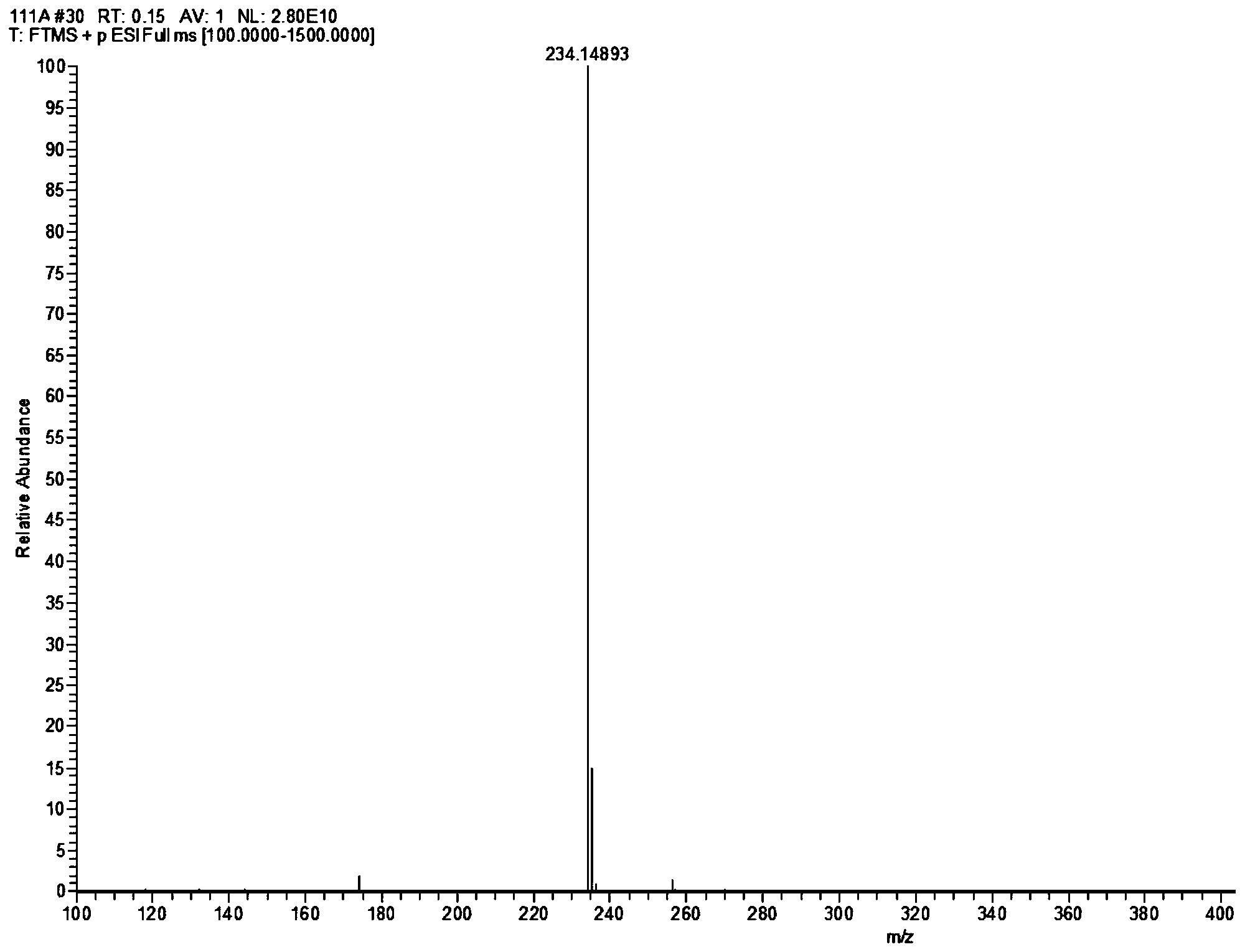
图1为本发明化合物13的高分辨质谱图;
图2为本发明化合物14的高分辨质谱图;
图3为本发明天然产物一枝蒿碱G的高分辨质谱图;
图4为本发明化合物15的高分辨质谱图;
图5为本发明化合物13的旋光值图;
图6为本发明化合物14的旋光值图;
图7为本发明天然产物一枝蒿碱G的旋光值图;
图8为本发明化合物15的旋光值图;
图9为本发明化合物13和化合物14的实验ECD和计算ECD谱,其中曲线A为化合物13的实验ECD曲线;曲线B为化合物14的实验ECD曲线;曲线C为化合物14的计算ECD曲线;
图10为本发明天然产物一枝蒿碱G和化合物15的实验ECD和计算ECD谱,其中曲线A为化合物15的实验ECD曲线;曲线B为一枝蒿碱G的实验ECD曲线;曲线C为一枝蒿碱G的计算ECD曲线。
具体实施方式
为了使本发明更加清楚,以下结合实施例,对本发明进行进一步详细说明,应当理解,所描述的实施例仅用于解释本发明,并不限定本发明。
实施例1
a、将化合物1为2-甲基-5-溴吡啶2.0g,(12mmol)溶于二氯甲烷溶液30ml,冰浴下分批加入纯度为85%的间氯过氧苯甲酸2.9g,(13.9mmol)后自然升温至室温,搅拌过夜反应,反应结束后将反应液倒入到饱和的亚硫酸钠溶液中淬灭,搅拌1个小时之后用二氯甲烷150ml萃取3次,合并有机相,用无水硫酸镁干燥,除去有机溶剂后得到氮氧化产物化合物2为5-溴-2-甲基-N-氧吡啶,产率93%,2.0g;
b、在氮气保护下,将化合物2为5-溴-2-甲基-N-氧吡啶1.0g(5.4mmol)溶于乙腈溶液20ml中,依次加入三甲基氰硅烷2.1g,(21.6mmol)和三乙胺2.2ml,(16.2mmol),回流12小时后,除去有机溶剂,用中压制备色谱为COMBIFLASH,流动相:体积比1:10的乙酸乙酯:石油醚纯化,得到化合物3为3-溴-6-甲基-2-氰基吡啶,产率85%,0.9g;
1H NMR(400MHz,Chloroform-d)δ7.98(d,J=8.2Hz,1H),7.24(d,J=3.0Hz,1H),2.57(s,3H);
c、真空状态下,将质量比40:60含结晶水的氯化锌7.2g(32.5mmol)加热至熔融除水,在氮气保护下,依次加入1,2-二氯乙烷75ml、化合物3为3-溴-6-甲基-2-氰基吡啶5.5g(27.9mmol)、丙二酸单乙酯钾盐11g(64.7mmol)和二异丙基乙胺1.5ml(9.1mmol),搅拌回流12小时,等体系冷却至室温后加入6N盐酸20ml,再次回流1小时,分出有机相,水相用二氯甲烷萃取3次,合并有机相后用无水硫酸镁干燥,除去有机溶剂,纯化,得到化合物4为3-(3-溴-6-甲基吡啶)-3-羰基丙酸乙酯,产率82%,6.6g;
1H NMR(400MHz,Chloroform-d)δ7.85(d,J=8.2Hz,1H),7.13(d,J=8.2Hz,1H),4.15(q,J=7.1Hz,2H),4.10(s,2H),2.51(s,3H),1.21(t,J=7.1Hz,4H);
d、将化合物4为3-(3-溴-6-甲基吡啶)-3-羰基丙酸乙酯1g,(3.5mmol)溶解在无水乙醇10ml中,冰浴中加入乙醇钠0.6g(3.9mmol),搅拌的同时加入3-溴丙烯0.7ml(3.9mmol),室温过夜反应,反应结束之后,除去有机溶剂,柱层析(流动相:体积比1:10的乙酸乙酯:石油醚)纯化,得到化合物5为2-(3-溴-6-甲基吡啶-2-甲酰基)-4-烯戊酸乙酯,产率97%,1.1g;
1H NMR(400MHz,Chloroform-d)δ7.85(d,J=8.3Hz,1H),7.12(d,J=8.2Hz,1H),5.92–5.79(m,1H),5.11(d,J=17.1,1.6Hz,1H),5.03(d,J=10.3,1.8Hz,1H),4.64(t,J=7.2Hz,1H),4.10(q,J=7.2Hz,2H),2.81–2.65(m,2H),2.52(s,3H),1.15(t,J=7.1Hz,3H);
e、将化合物5为2-(3-溴-6-甲基吡啶-2-甲酰基)-4-烯戊酸乙酯1g(3.1mmol)溶于体积比3:1的二氧六环和水的混合溶液中,加入四三苯基膦钯催化剂0.4g(0.3mmol)、碳酸钠1g(9.3mmol)和异丙烯基硼酸频哪醇酯0.71ml(3.3mmol),搅拌加热回流3小时,待体系冷却至室温后除去溶剂,柱层析(流动相:体积比1:10的乙酸乙酯:石油醚)纯化,得到化合物6为2-(3-异丙烯基-6-甲基吡啶-2-甲酰基)-4-戊烯酸乙酯,产率92%,0.8g;
1H NMR(400MHz,Chloroform-d)δ7.45(d,J=7.9Hz,1H),7.22(d,J=7.9Hz,1H),5.93–5.80(m,1H),5.15–5.04(m,1H),5.00(d,J=10.0Hz,1H),4.83(d,J=1.8Hz,1H),4.72(t,J=7.2Hz,1H),4.15–4.06(m,2H),2.80–2.63(m,2H),2.55(s,3H),2.03(d,J=1.4Hz,3H),1.14(td,J=7.1,0.9Hz,3H);
f、在氮气保护下,将化合物6为2-(3-异丙烯基-6-甲基吡啶-2-甲酰基)-4-戊烯酸乙酯1g(3.5mmol)溶于干燥二氯甲烷溶液30ml中,加入Grubbs二代催化剂0.3g(0.3mmol),加热回流反应12小时,反应结束后,待体系冷却至室温,除去有机溶剂,柱层析(流动相:体积比1:5的乙酸乙酯:石油醚),得到化合物7为2,5-二甲基-5-烯-9-氧代-环庚烷并[b]吡啶-8-甲酸乙酯,产率53%,0.5g;
1H NMR(400MHz,Chloroform-d)δ12.45(s,1H),7.75(d,J=8.2Hz,1H),7.24(d,J=8.4Hz,1H),6.08(t,J=8.3,7.4Hz,1H),4.31(q,J=7.1Hz,2H),2.68(s,3H),2.52(d,J=6.8Hz,2H),2.09(s,3H),1.37(t,J=7.1Hz,3H)。13C NMR(101MHz,Chloroform-d)δ171.08,164.65,156.65,149.48,134.98,134.17,131.94,130.15,123.64,105.79,61.06,24.51,21.39,19.12,14.23;
g、在冰浴下,将化合物7为2,5-二甲基-5-烯-9-氧代-环庚烷并[b]吡啶-8-甲酸乙酯100mg(0.4mmol)溶于无水甲醇5ml中,加入硼氢化钠15mg(0.4mmol),体系自然升温至室温,反应3h后,加水淬灭反应,除去溶剂,柱层析(流动相:体积比1:3的乙酸乙酯:石油醚)纯化,得到化合物8为2,5-二甲基-5-烯-9-羟基-环庚烷并[b]吡啶-8-甲酸乙酯,产率78%,80mg;
1H NMR(400MHz,DMSO-d6)δ7.71(d,J=7.9Hz,1H),7.28(d,J=7.9Hz,1H),6.03–5.96(m,1H),5.40(d,J=5.4Hz,1H),4.81(t,J=5.9Hz,1H),4.06–3.88(m,2H),3.49(dt,J=10.8,6.6Hz,1H),2.52(s,3H),2.27–2.07(m,2H),2.04(s,3H),1.10(t,J=7.1Hz,3H);
h、将化合物8为2,5-二甲基-5-烯-9-羟基-环庚烷并[b]吡啶-8-甲酸乙酯100mg(0.4mmol)溶于吡啶10ml中,加热至温度60℃后加入MsCl45.6mg(0.4mmol),反应3小时后,冷却至室温,加水淬灭反应,再用二氯甲烷30ml萃取分出有机相,有机相用无水硫酸镁干燥,柱层析(流动相:体积比1:5的乙酸乙酯:石油醚)纯化,得到化合物9为2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸乙酯,产率87%,84mg;
1H NMR(400MHz,Chloroform-d)δ7.79(d,J=8.2Hz,1H),7.73(s,1H),5.84(t,J=7.2Hz,0H),4.27(q,J=7.1Hz,2H),2.67(d,J=7.2Hz,2H),2.62(s,3H),2.10(d,J=1.4Hz,3H),1.33(t,J=7.1Hz,3H)。
i、将化合物9为2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸乙酯100mg(0.4mmol)溶于体积比1:1的四氢呋喃和水的混合溶液中,加入氢氧化锂24mg(1mmol),室温搅拌过夜反应,反应结束后除去溶剂,所得的粗产品溶于无水甲醇中,冰浴下加入碘甲烷70.5mg(0.5mmol)和1,8-二氮杂双环[5.4.0]十一碳-7-烯125.5mg(0.5mmol),室温搅拌过夜反应,反应结束后除去溶剂,柱层析(流动相:体积比1:10的乙酸乙酯:石油醚)纯化,得到化合物10为2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸甲酯,总产率83%,76mg;
1H NMR(400MHz,Chloroform-d)δ7.78(d,J=8.2Hz,1H),7.71(s,1H),7.13(d,J=8.2Hz,1H),5.83(td,J=7.2,1.5Hz,1H),3.81(s,3H),2.66(d,J=7.3Hz,2H),2.60(s,3H),2.09(d,J=1.5Hz,3H)。
j、将化合物10为2,5-二甲基-5,8-二烯-环庚烷并[b]吡啶-8-甲酸甲酯50mg(0.2mmol)溶于无水甲醇10ml中,冰浴下加入浓度为0.5%的钯碳催化剂15mg,通入氢气,室温搅拌过夜反应,反应结束后除去溶剂,用薄层色谱层析方法分离纯化(展开剂:体积比1:5的乙酸乙酯:石油醚),分别得到化合物11为反-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基和化合物12为顺-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基;
化合物11(31mg):1H NMR(400MHz,Chloroform-d)δ7.34(d,J=7.6Hz,1H),6.95(d,J=7.9Hz,1H),3.63(s,2H),3.44(d,J=14.0Hz,1H),3.29(d,J=14.0Hz,1H),3.03–2.93(m,1H),2.72–2.63(m,1H),2.51(s,3H),2.19–2.07(m,1H),2.02–1.92(m,1H),1.81–1.70(m,2H),1.31(d,J=7.3Hz,3H);
化合物12(10.6mg):1H NMR(400MHz,Methanol-d4)δ7.58(d,J=8.0Hz,1H),7.14(d,J=8.0Hz,1H),3.70(s,3H),3.29–3.27(m,1H),3.24–3.17(m,1H),3.06(p,J=7.3Hz,1H),2.51–2.48(m,1H),2.47(s,3H),2.45–2.38(m,1H),2.20–2.10(m,1H),2.06–1.96(m,1H),1.96–1.88(m,1H),1.36(d,J=7.1Hz,3H);
k、将化合物11为反-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基和化合物12为顺-(5-甲基-8-甲酸甲酯)-环庚烷并[b]吡啶-2-甲基分别用半制备型高效液相色谱(SHIMADZU LC-20A),手型柱CHIRALPAK ID(Lot No.ID00CE-QI011)拆分,对映异构体(流动相:体积比98:2的正己烷:乙醇),得到化合物13为(5S,8R)-一枝蒿碱G;化合物14为(5R,8S)-一枝蒿碱G;化合物15为(5R,8R)-一枝蒿碱G;一枝蒿碱G。
实施例2
将一枝蒿碱G(Rupestine G)和其三个异构体化合物13为(5S,8R)-一枝蒿碱G;化合物14为(5R,8S)-一枝蒿碱G;化合物15为(5R,8R)-一枝蒿碱G用美国AB SCIEX公司Qstar Elite四极杆-飞行时间杂交高分辨质谱仪测定其精确分子量,用Rudolph RS Autopol VI automatic polarimeter测定旋光值,用Chirascan圆二色谱仪实验测定EDC和用德国COSMOlogic GmbH&Co.KG TmoleX 3.4软件计算ECD谱比较确定其绝对构型,一枝蒿碱G(Rupestine G)和其异构体的高分辨质谱见图1-图4。
一种天然产物(±)-一枝蒿碱G的全合成及对映异构体的拆分方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0