

IPC分类号 : C12N15/54,C12N9/10,C12N1/21,C12P25/00,C12R1/125







专利摘要
本发明公开了枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF及应用,枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF序列用SEQIDNo.1所示。本发明所构建的包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌生物安全,遗传背景清晰,可以大幅提高枯草芽孢杆菌合成核黄素的能力,提高核黄素积累水平20%以上。
权利要求
1.枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF,其特征是所述突变基因purF序列用SEQ ID No.1所示。
2.权利要求1所述的枯草芽孢杆菌突变基因purF所编码的氨基酸序列,其特征是所述氨基酸序列用SEQ ID No.2所示。
3.包含有权利要求1枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌。
4.权利要求3所述的工程菌在产核黄素的应用。
说明书
技术领域
本发明属于生物技术与分子生物学领域,具体地涉及枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF及该突变基因编码的氨基酸序列,包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌及该菌在产核黄素的应用。
背景技术
以细菌为宿主菌的基因工程菌具有发酵周期短、原料要求简单、成熟的基因工程技术等优点。在芽孢杆菌属中,包括枯草芽孢杆菌(Bacillus subtilis)在内的许多菌株具有可靠的安全性。传统菌株选育发现,枯草芽孢杆菌的突变株能够过量合成叶酸、腺苷、肌苷、鸟苷、核黄素等一系列嘌呤途径代谢中间产物或该途径的衍生代谢产物,成为选育高产核苷类代谢产物重要出发菌株。枯草芽孢杆菌作为模式菌株,对其生理生化特性及遗传背景已经有了比较深入的了解,相关的分子生物学方法和基因操作技术都比较成熟,有利于通过理性代谢工程及系统生物学手段来选育核苷类代谢产物(如核黄素)高产菌种。核黄素(分子式C17H20O6N4,IUPAC中文名:7,8-二甲基-10-(1'-D-核糖基)-异咯嗪)是人体必需的13种维生素之一,是黄素酶类的辅酶组成部分,在生物体内主要以黄素单核苷酸(FMN)和黄素腺嘌呤二核苷酸(FAD)的形式存在。它作为黄素蛋白的辅酶参与机体组织呼吸链电子传递及氧化还原反应,在呼吸和生物氧化中起着重要的作用。
枯草芽孢杆菌的嘌呤合成途径包括10步反应,其中催化这10步反应所需要的酶由嘌呤操纵子负责编码。嘌呤核苷酸生物合成的第一步是由磷酸戊糖焦磷酸激酶催化,5'-磷酸-D-核糖与ATP反应生成5'-磷酸核糖焦磷酸(PRPP),随后经过9步反应生成肌苷酸(IMP),反应生成的IMP并不堆积在细胞内,而是迅速转变为腺苷酸(AMP)和鸟苷酸(GMP)。嘌呤从头合成是合成嘌呤核苷酸的主要途径,此过程要消耗氨基酸及ATP,同时嘌呤从头合成途径受到严格的多重调控机制。
嘌呤操纵子存在两种相互独立的代谢调控机制:腺嘌呤介导的转录起始阻遏机制和鸟嘌呤通过作用于前导mRNA来调节其转录的衰减机制。嘌呤基因转录起始位点上游-145~-29区的顺式作用元件是阻遏转录起始的调节蛋白结合位点。该调控区域的缺失突变会使腺嘌呤的阻遏作用消失,但对鸟嘌呤的衰减机制几乎没有影响。腺嘌呤介导的转录起始阻遏机制利用属于LacI类的调节蛋白-PurR阻抑物调节嘌呤基因的表达。PurR蛋白的调控是通过胞内代谢物PRPP介导的,PurR蛋白与DNA的结合不受腺嘌呤、腺苷或腺苷酸的影响,但PRPP可抑制它们的结合。嘌呤合成途径中除了转录水平的调节,还存在着终产物对关键酶的反馈抑制作用,对合成速度有着精细的调节,不仅调节嘌呤核苷酸的总量,而且使ATP和GTP的水平保持相对平衡。PRPP转酰胺酶是嘌呤从头合成途径的关键调节酶,该酶活的高低直接影响进入嘌呤合成途径的通量。PRPP转酰胺酶受到ATP,AMP,GTP和GMP的反馈抑制作用。解除嘌呤途径受到的反馈抑制对于提高嘌呤途径的通量以及核苷类(肌苷和鸟苷)代谢产物或源于嘌呤途径代谢物的积累具有重要的意义。
目前,尚未有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF用于核黄素高产菌种的选育的报道。
发明内容
本发明的目的是提供一种能够解除嘌呤核苷类反馈抑制的枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF。
本发明的第二个目的是提供一种枯草芽孢杆菌突变基因purF所编码的氨基酸序列。
本发明的第三个目的是提供一种包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌。
本发明的第四个目的是提供一种包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌的应用。
本发明的技术方案概述如下:
枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF,所述突变基因purF序列用SEQ ID No.1所示。
枯草芽孢杆菌突变基因purF所编码的氨基酸序列,所述氨基酸序列用SEQ ID No.2所示。
包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌。
包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌在产核黄素的应用。
本发明所构建的包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌生物安全,遗传背景清晰,可以大幅提高枯草芽孢杆菌合成核黄素的能力,提高核黄素积累水平20%以上。
附图说明
图1为无痕基因操作所需pSS质粒图谱。
图2为无痕基因操作所需purF点突变引入的pSS-purF*-FB质粒图谱。
图3为牛血清蛋白BSA标准曲线绘制。
图4为PRPP转酰胺酶野生酶(B.subtilis RC2)和突变酶(B.subtilis RC5)酶活性测定。
图5为不同浓度ATP对PRPP转酰胺酶野生酶(B.subtilis RC2)和突变酶(B.subtilis RC5)酶活性的抑制作用。
图6为不同浓度AMP对PRPP转酰胺酶野生酶(B.subtilis RC2)和突变酶(B.subtilis RC5)酶活性的抑制作用。
图7为不同浓度GTP对PRPP转酰胺酶野生酶(B.subtilis RC2)和突变酶(B.subtilis RC5)酶活性的抑制作用。
图8为不同浓度GMP对PRPP转酰胺酶野生酶(B.subtilis RC2)和突变酶(B.subtilis RC5)酶活性的抑制作用。
图9为菌株B.subtilis RC2和B.subtilis RC5核黄素合成水平发酵验证。
具体实施方式
下面结合实施例对本发明做进一步说明,下述实施例是为了使本领域的技术人员能够更好地理解本发明,但对本发明不作任何限制。
本发明所用到的原始菌株B.subtilis168来源为BGSC(Bacillus Genetic Stock Center,http://www.bgsc.org/)。本发明所用到的原始质粒pUC18购买于生工生物工程(上海)股份有限公司(http://www.sangon.com/)。
本发明所用到的ATP,AMP,GTP,GMP和PRPP(磷酸核糖焦磷酸)药品从Sigma公司(http://www.sigmaaldrich.com/sigma-aldrich)购买,所用限制性内切酶、去磷酸化酶、DNA连接酶等分子生物学试剂从Thermo公司购买(http://www.thermoscientificbio.com/fermentas),所用其他生化试剂从生工生物工程(上海)股份有限公司购买(http://www.sangon.com/)。
实施例1:基础操作质粒pSS的构建
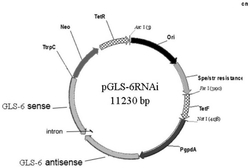
利用PCR反应以pC194(来源:Bacillus Genetic Stock Center,BGSC,http://www.bgsc.org/)质粒为模版使用上下游引物pSS-P1和pSS-P2获取cat基因、以B.subtilis168基因组为模版使用上下游引物pSS-P3和pSS-P4获取upp基因,再以上面的两个片段为模版,利用融合PCR反应使用上下游引物pSS-P1和pSS-P4获取重组片段Cat-Upp。将该重组片段与pUC18(通用载体)质粒经酶切、酶连、转化、验证等操作后,得到基础操作质粒pSS,见图1。
本发明对该基础操作质粒pSS的构建方法以及抗性基因的选择不做限定。
实施例2:purF点突变引入质粒pSS-purF*-FB构建
利用purF-F-U和purF-F-L一对引物,以B.subtilis168基因组为模版,使用KOD-plus高保真DNA聚合酶扩增得到大小为836bp的上游同源臂purF*-F,其中下引物purF-F-L中引入突变点D293V。purF*-F的PCR片段经切胶回收后,使用Thermo Fast digest BglII和XhoI双酶切,连接、转化质粒pSS后得到质粒pSS-purF*-F。
利用purF-B-Fsn-1和purF-B-Fsn-2一对引物,以B.subtilis168基因组为模版,使用KOD-plus高保真DNA聚合酶扩增得到大小为345bp的片段A,其中上引物purF-B-Fsn-1中引入突变点K316Q,下引物purF-B-Fsn-2中引入突变点S400W。利用purF-B-Fsn-3和purF-B-Fsn-4一对引物,以B.subtilis168基因组为模版,使用KOD-plus高保真DNA聚合酶扩增得到大小为594bp的片段B,其中上引物purF-B-Fsn-3中引入突变点S400W。以片段A和片段B为模版,利用融合PCR反应使用上下游引物purF-B-U和purF-B-L获取大小为818bp的下游同源臂purF*-B。purF*-B的PCR片段经切胶回收后,使用Thermo Fast digest SalI和KpnI双酶切,连接、转化质粒pSS-purF*-F后得到质粒pSS-purF*-FB。质粒构建成功后送测序检查突变点成功引入至质粒载体中,见图2。
实施例3:枯草芽孢杆菌系统出发菌株B.subtilis BUK的构建
本发明中所用到的枯草芽孢杆菌出发菌株B.subtilis BUK来源于B.subtilis168,详细构建过程可参考公开发表文献1。
该菌株具有诱导条件下快速制备感受态细胞的能力,并同时具有较高的外源DNA的吸收能力。
实施例4:工程菌株B.subtilis RC1和B.subtilis RC2构建过程
(1)向菌株B.subtilis BUK基因组上无痕引入基因突变ribC(G596A)操作流程如下:
利用ribC-F-U、ribC-F-L和ribC-B-U、ribC-B-L两对引物,以B.subtilis168为模版,使用KOD-plus高保真DNA聚合酶扩增分别得到大小为894bp和928bp的上下游同源臂ribC*-F和ribC*-B。ribC*-F的PCR片段经切胶回收后,使用Thermo Fast digest AatII和XhoI双酶切,连接、转化质粒pSS后得到质粒pSS-ribC*-F。ribC*-B的PCR片段经切胶回收后,使用Thermo Fast digest SalI和ScaI双酶切,连接、转化质粒pSS-ribC*-F后得到这质粒pSS-ribC*-FB。
将测序结果正确的质粒pSS-ribC*-FB通过Spizizen转化导入枯草芽孢杆B.subtilis BUK中,用含5μg/mL氯霉素LB固体培养基中筛选重组成功的阳性克隆,并用菌落PCR验证。挑出的转化子接种于5ml LB液体培养基中,200rpm震荡培养6h(OD约为2),并在五氟尿嘧啶基本培养基(添加终浓度为5μmol/L FMN)上挑出菌落,使用引物ribC-F-U、ribC-B-L进行PCR和测序验证,得到ribC(G596A)正确引入的阳性菌株B.subtilis RC1。
(2)向菌株B.subtilis RC1基因组上无痕引入基因突变ribO(G+39A)具体操作如下:
利用ribO-F-U、ribO-F-L和ribO-B-U、ribO-B-L两对引物,以B.subtilis168为模版,使用KOD-plus高保真DNA聚合酶扩增分别得到大小为922bp和1234bp的上下游同源臂ribO*-F和ribO*-B。ribO*-F的PCR片段经切胶回收后,使用Thermo Fast digest AatII和XhoI双酶切,连接、转化质粒pSS后得到质粒pSS-ribO*-F。ribO*-B的PCR片段经切胶回收后,使用Thermo Fast digest SalI和KpnI双酶切,连接、转化质粒pSS-ribO*-F后得到质粒pSS-ribO*-FB。
将测序结果正确的质粒pSS-ribO*-FB通过Spizizen转化导入枯草芽孢杆B.subtilis RC1中,用含5μg/mL氯霉素LB固体培养基中筛选重组成功的阳性克隆,并用菌落PCR验证。挑出的转化子接种于5ml LB液体培养基中,200rpm震荡培养6h(OD约为2),并在五氟尿嘧啶基本培养基(添加终浓度为5μmol/L FMN)上挑出菌落,使用引物ribO-F-U、ribO-B-L进行PCR和测序验证,得到ribO(G+39A)正确引入的阳性菌株B.subtilis RC2。
实施例5:工程菌株B.subtilis RC5(含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌)构建
将实施例2中测序结果正确的质粒pSS-purF*-FB通过Spizizen转化导入枯草芽孢杆B.subtilis RC2中,用含5μg/mL氯霉素LB固体培养基中筛选重组成功的阳性克隆。将正确的阳性克隆接种于5mL LB液体培养基中,200rpm震荡培养6h,并在五氟尿嘧啶基本培养基上培养并挑出菌落,使用引物purF-F-U、purF-B-L进行PCR验证和测序验证,得到purF正确引入的阳性菌株,即含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌,命名为B.subtilis RC5,其中枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF(D293V,K316Q,S400W)和枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF所编码的氨基酸序列分别用SEQID No.1和SEQ ID No.2所示。
LB液体培养基配方为:10g/L蛋白胨,5g/L酵母提取物,10g/L NaCl,调节pH至7.5。0.1Mpa压力下灭菌20min。
LB固体培养基配方为:在LB液体培养基中加入琼脂粉(终浓度15g/L),0.1Mpa压力下灭菌20min。
五氟尿嘧啶基本培养基配方见表1:
表1五氟尿嘧啶基本培养基配方
其中,10×Spizizen基本培养基:2g/L(NH4)2SO4,18.3g/L K2HPO4,6g/L KH2PO4,12g/L柠檬酸钠,pH7.2,0.1Mpa压力下灭菌20min。
1000×微量元素:27g/L FeCl3·6H2O,2g/L ZnCl2·4H2O,2g/L CaCl2·2H2O,2g/LNa2MoO4·2H2O,1.9g/L CuSO4·5H2O,0.5g/L H3BO3,pH7.2,0.1Mpa压力下灭菌20min。
实施例6:PRPP转酰胺酶野生酶与突变酶活性测定
1、粗酶液的制备过程通过无菌条件分别吸取LB液体培养基培养至对数生长期(OD600=1.0)的枯草芽孢杆菌B.subtilis RC2和B.subtilis RC5菌液10mL,迅速将菌液冷冻离心,用pH=7.0的磷酸缓冲液重悬离心两次后保存菌体,用500μL pH=7.0的磷酸缓冲液重悬菌体,用细胞破碎仪破碎10min后8000rpm冷冻离心10min,吸取上清即为粗酶液。磷酸缓冲液:10.9g/L Na2HPO4,2.3g/L Na2HPO4,调节pH至7.0,0.1Mpa压力下灭菌20min。
2、Bradford法测定粗酶液浓度Bradford法定量蛋白质浓度是根据蛋白质与染料相结合的原理设计的。该法的原理是考马斯亮蓝G-250染料在酸性溶液中与蛋白质结合,使得染料的最大吸收峰从465nm变为595nm,溶液的颜色也由棕黑色变为蓝色。蛋白-染料复合物具有高的消光系数,因此大大提高蛋白质测量的灵敏度,最低检出量为1μg蛋白。采用牛血清蛋白(BSA)作为标准蛋白,其标准曲线见图3。测量方法分为以下两步骤:
(1)取5支试管,按表2加入试剂;
表2标准曲线测定体系
(2)混匀后静置2min,以0号管作空白对照,测定各管595nm下的OD值,以OD值为纵坐标,蛋白浓度为横坐标作图,得到标准曲线(图3)。标准曲线的公式:y=0.013x+0.1175(R2=0.998)
(3)粗酶液中蛋白质浓度的测定:将未知浓度的粗酶液(通过适当浓度的稀释,使其浓度控制在10~100μg/mL范围内)加到试管内,再加入考马斯亮蓝G-250溶液5ml混匀,测其595nm下的OD值,对照图3所示标准曲线求出粗酶液中蛋白液的浓度。
3、PRPP转酰胺酶野生酶与突变酶酶活测定
PRPP转酰胺酶催化如下反应:L-谷氨酰胺+PRPP+H2O=PRA+L-谷氨酸。具体步骤如下:
(1)取一只试管进行酶促反应,加入以下试剂:2.5mM PRPP,200mM L-谷氨酰胺,10mMMgCl2,1mg/mL牛血清蛋白,50mM Tris-HCl(pH=8.0),100ng步骤1获得的粗酶液,总体积用双蒸水补至100μL。
(2)将酶促反应体系置于37℃反应30min,然后于100℃放置1min终止反应。
(3)利用SBA生物传感器测定L-谷氨酸浓度,本实验利用测定产物L-谷氨酸的生成量来确定酶活力的大小,即在特定的条件下,1min内1mg粗酶液催化生成L-谷氨酸的含量(单位:μmol/min/mg粗蛋白),从而计算PRPP转酰胺酶野生酶与突变酶的酶活力(图4)。
从酶活力结果可以看出,相对于B.subtilis RC2菌株野生型PRPP转酰胺酶的酶活力,B.subtilis RC5菌株提取的PRPP转酰胺酶突变酶的酶活力提高了1.63倍。
实施例7:不同浓度ATP,AMP,GTP和GMP对PRPP转酰胺酶野生酶和突变酶酶活性的抑制作用。
ATP,AMP,GTP和GMP对PRPP转酰胺酶野生酶有抑制作用,在酶促反应体系中添加不同浓度梯度的ATP,AMP,GTP或GMP来检测PRPP转酰胺酶突变酶的酶活力,考察突变基因purF编码的PRPP转酰胺酶突变酶是否解除了反馈抑制效应。
配制酶促反应体系:2.5mM PRPP,200mM L-谷氨酰胺,10mM MgCl2,1mg/mL牛血清蛋白,50mM Tris-HCl(pH=8.0),100ng步骤1获得的粗酶液,总体积用双蒸水补至100μL。
向酶促反应体系中分别添加浓度为0mM,10mM,20mM,40mM的ATP,按照实施例6中酶活性检测方法检测PRPP转酰胺酶野生酶(用B.subtilis RC2表示)与突变酶(用B.subtilis RC5表示)酶活力变化趋势,实验结果见图5。从酶活力变化趋势结果可以看出,随着ATP添加浓度的增加,B.subtilis RC2菌株提取的PRPP转酰胺酶野生酶酶活力不断下降,PRPP转酰胺酶野生酶受到ATP的反馈抑制;而B.subtilis RC5菌株提取的PRPP转酰胺酶突变酶在低浓度的ATP条件下酶活力提高,同时也缓解了高浓度ATP条件下的反馈抑制作用。
在酶促反应体系中分别添加浓度为0mM,1mM,10mM,40mM的AMP,按照实施例6中酶活性检测方法检测PRPP转酰胺酶野生酶(用B.subtilis RC2表示)与突变酶(用B.subtilis RC5表示)酶活力变化趋势,实验结果见图6。从酶活力变化趋势结果可以看出,随着AMP添加浓度的增加,B.subtilis RC2菌株提取的PRPP转酰胺酶野生酶酶活力快速下降,PRPP转酰胺酶野生酶受到AMP严格的反馈抑制;而B.subtilis RC5菌株提取的PRPP转酰胺酶突变酶缓解了AMP反馈抑制作用。
在酶促反应体系中分别添加浓度为0mM,10mM,20mM,40mM的GTP,按照实施例6中酶活性检测方法检测PRPP转酰胺酶野生酶(用B.subtilis RC2表示)与突变酶(用B.subtilis RC5表示)酶活力变化趋势,实验结果见图7。从酶活力变化趋势结果可以看出,随着GTP添加浓度的增加,B.subtilis RC2菌株提取的PRPP转酰胺酶野生酶酶活力下降,PRPP转酰胺酶野生酶受到GTP严格的反馈抑制;而B.subtilis RC5菌株提取的PRPP转酰胺酶突变酶在低浓度的GTP条件下酶活力提高,同时解除了高浓度GTP条件下的反馈抑制作用。
在酶促反应体系中分别添加浓度为0mM,1mM,10mM,40mM的GMP,按照实施例6中酶活性检测方法检测PRPP转酰胺酶野生酶(用B.subtilis RC2表示)与突变酶(用B.subtilis RC5表示)酶活力变化趋势,实验结果见图8。从酶活力变化趋势结果可以看出,随着GMP添加浓度的增加,B.subtilis RC2菌株提取的PRPP转酰胺酶野生酶酶活力快速下降,PRPP转酰胺酶野生酶受到GMP严格的反馈抑制;而B.subtilis RC5菌株提取的PRPP转酰胺酶突变酶缓解了GMP反馈抑制作用。
实施例8:工程菌株B.subtilis RC2和B.subtilis RC5核黄素合成能力对比
分别将菌株B.subtilis RC2和B.subtilis RC5在含100g/L葡萄糖的YE培养基中,500mL锥形瓶装液量为50mL,摇床转速240rpm,41℃培养60h。接种方式为:LB平板活化菌株,37℃过夜培养,挑单菌落接种5mL LB培养基的摇管,培养12h后,按初始OD为0.02接种量转YE培养基。
YE培养基配方:100g/L葡萄糖,20g/L酵母粉,0.5g/L MgSO4,0.5g/L KH2PO4,0.5g/LK2HPO4,调节pH至7.0,0.1Mpa压力下灭菌20min。
发酵结果见图9。从发酵结果可以看出,相对于出发菌株B.subtilis RC2,包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌(B.subtilis RC5)提高了核黄素产量20%以上。说明这种遗传修饰对于核黄素等核苷类高产菌株选育具有较好的应用前景。
本发明的菌株的构建,其步骤的前后顺序不限定,本领域的技术人员按本发明公开的内容达到本发明的目的均属于本发明的保护范围。
本发明中的菌株代号如B.subtilis RC2,B.subtilis RC5等是为了方便描述,但不应理解为对本发明的限定。
上述方法构建的包含有枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF的工程菌的用途,包括但是不局限于核黄素。
参考文献1:Shi,T.,Wang,G.,Wang,Z.,Fu,J.,Chen,T.,Zhao,X.,2013.Establishment of a Markerless Mutation Delivery System in Bacillus subtilis Stimulated by a Double-Strand Break in the Chromosome.PLoS one.8,e81370.
表3菌株构建所用引物序列
枯草芽孢杆菌编码PRPP转酰胺酶突变基因purF及应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0