









专利摘要
本发明公开了一种Ivosidenib及其中间体的手性合成方法,该方法包括:先加入L‑焦谷氨酸衍生物、邻氯苯甲醛、3‑氨基‑5‑氟吡啶、手性质子酸催化剂和溶剂搅拌,再加入1,1‑二氟‑3‑异环丁腈反应,反应液经纯化,得到Ivosidenib或其中间体。本发明的合成方法反应避免了手性柱纯化,适合工业化规模生产,去除了安全性存疑的溶剂,制备的药品安全性高,在药品生产中具有重要的应用意义。
权利要求
1.一种Ivosidenib及其中间体的手性合成方法,其特征在于,包括:
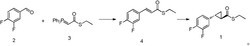
先加入L-焦谷氨酸衍生物、邻氯苯甲醛、3-氨基-5-氟吡啶、手性质子酸催化剂和溶剂搅拌,再加入1,1-二氟-3-异环丁腈反应,反应液经纯化,得到Ivosidenib或其中间体,反应方程式如式I所示:
R为H或4-氰基吡啶-2-基。
2.根据权利要求1所述的Ivosidenib及其中间体的手性合成方法,其特征在于,反应是L-焦谷氨酸衍生物、邻氯苯甲醛、3-氨基-5-氟吡啶和1,1-二氟-3-异环丁腈的4组分Ugi反应。
3.根据权利要求1所述的Ivosidenib及其中间体的手性合成方法,其特征在于,所述手性质子酸催化剂为具有结构式Ⅰ-Ⅲ的手性质子酸:
4.根据权利要求3所述的Ivosidenib及其中间体的手性合成方法,其特征在于,手性质子酸的R
5.根据权利要求4所述的Ivosidenib及其中间体的手性合成方法,其特征在于,R
6.根据权利要求3所述的Ivosidenib及其中间体的手性合成方法,其特征在于,所述的手性质子酸为以下具体化合物中的一种:
7.根据权利要求1所述的Ivosidenib及其中间体的手性合成方法,其特征在于,反应适用的温度范围为-80℃~80℃。
8.根据权利要求1所述的Ivosidenib及其中间体的手性合成方法,其特征在于,反应的溶剂为烃类、醇类、醚类、酯类或腈类溶剂。
9.根据权利要求8所述的Ivosidenib及其中间体的手性合成方法,其特征在于,反应的溶剂为烃类溶剂;
烃类溶剂包括直链烃类、环烃类、芳烃类以及相关卤代烃溶剂。
10.根据权利要求9所述的Ivosidenib及其中间体的手性合成方法,其特征在于,所述的烃类溶剂为二氯甲烷或二氯乙烷。
说明书
技术领域
本发明涉及医药化工领域,具体的说,涉及一种Ivosidenib及其中间体的手性合成方法。
背景技术
Ivosidenib是Agios Pharms公司开发的一种靶向突变的异枸橼酸脱氢酶-1(IDH1)抑制剂,化学名称:(2S)-N-[(1S)-1-(2-氯苯基)-2-[(3,3-二氟环丁基)氨基]-2-氧代乙基]-N-(5-氟吡啶-3-基)-1-(4-氰基吡啶-2-基)-5-氧代吡咯烷-2-甲酰胺。该药于2018年7月在美国批准上市,用于存在易感IDH1突变的复发性或难治性急性髓系白血病(AML)成人患者的治疗。
Ivosidenib上市不久,其合成方法文献报道极少,关键步骤和化合物专利US2013190249A1报道完全一致(同时参见文献《中国药物化学杂志》2019,29(2),163和《中国医药工业杂志》2019,50(1),1-33),即L-焦谷氨酸、邻氯苯甲醛、3-氨基-5-氟吡啶和1,1-二氟-3-异环丁腈在三氟乙醇中发生4组分Ugi反应,得到混旋的Ivosidenib中间体A经Buchwald-Hartwig反应得到混旋的Ivosidenib产品,经手性柱拆分得到Ivosidenib,方程式如下:
然而,上述工艺存在的缺点是:一、产品需经手性柱拆分,工艺无法大规模工业化;二、Ugi反应的溶剂为三氟乙醇,不仅仅是溶剂成本高,而且三氟乙醇毒性未明,应该归属为ICH溶剂分类中的4类溶剂,即无毒理学数据,应当审慎使用的溶剂,可能存在药品安全风险。
发明内容
本发明的目的在于,针对现有技术合成Ivosidenib存在的缺点,提供一种新的Ivosidenib及其中间体的合成方法,该合成方法为手性4组分Ugi合成方法,无需手性柱拆分,避免采用三氟乙醇溶剂。
为解决上述技术问题,本发明的技术方案包括:
在手性质子酸催化剂催化下的手性4组分Ugi反应:先加入L-焦谷氨酸衍生物、邻氯苯甲醛、3-氨基-5-氟吡啶、手性磷酸催化剂和溶剂搅拌,再加入1,1-二氟-3-异环丁腈反应,反应液经标准方法纯化,得到Ivosidenib或其中间体,反应方程式如式I所示:
本发明中,反应是L-焦谷氨酸衍生物、邻氯苯甲醛、3-氨基-5-氟吡啶和1,1-二氟-3-异环丁腈的4组分Ugi反应。
本发明中,L-焦谷氨酸衍生物为L-焦谷氨酸或N-(4-氰基吡啶-2-基)L-焦谷氨酸。
本发明中,手性质子酸为具有结构式Ⅰ-Ⅲ的手性质子酸,优选Ⅲ型手性质子酸。
本发明中,手性质子酸的R1取代基为氢、溴或氰基,R2取代基为氢、苯基或取代苯基,其中,苯基上的取代基为烷基,所述的烷基包括链烷基或者环烷基,进一步优选为C1~C6链烷基或者C1~C6环烷基;R3取代基为羟基或三氟磺酰胺基,优选R1取代基为氰基,R2取代基为2,4,6-三异丙基苯基或2,4,6-三环己基苯基,R3取代基为三氟磺酰胺基。
进一步地,所述的手性质子酸为以下具体化合物中的一种:
本发明中,反应适用的温度范围为-80℃~80℃,优选-30℃~30℃。
本发明中,反应的溶剂为烃类、醇类、醚类、酯类或腈类溶剂,优选烃类溶剂;烃类溶剂包括直链烃类、环烃类、芳烃类以及相关卤代烃溶剂。所述的烃类溶剂进一步优选二氯甲烷或二氯乙烷。
同现有技术相比,本发明的有益效果体现在:
(1)本发明通过采用手性质子酸作为催化剂进行反应,同时,反应一步进行,可以将消旋的Ugi反应和手性柱拆分二步缩短到手性Ugi反应一步,规避了手性柱拆分不易大规模工业化的限制。
(2)Ugi反应去除了ICH4类溶剂三氟乙醇,提高了药品的安全性。
具体实施方式
参照下列实施例说明本发明的特定实施方案。这些实施例是用以阐明本发明,而非以任何方式限制本发明。
催化剂的制备参考文献分别参考《Journal of the American ChemicalSociety》2018,140,10374-10381、2019,141,8,3414-3418及其参考文献报道。原料N-(4-氰基吡啶-2-基)L-焦谷氨酸的合成参考化合物专利US2013190249A1中Buchwald-Hartwig反应步骤合成。
产品de值的液相色谱检测条件:
仪器:Waters E2695液相色谱仪
色谱柱:CHIRALCEL ID 4.6mm×250mm×5μm
流动相:正己烷/异丙醇80/20
检测波长:254nm
柱温:35℃
流速:1.0mL/min
实施例1 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(5mmol,1.57g)和乙腈(10mL),20℃左右搅拌30分钟,升温到内温80℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。旋干溶剂,加入二氯甲烷(20mL)溶解,用5%冰的碳酸钠溶液(30mL)洗涤有机相2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为49%,液相检测de值为12%。
实施例2 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(5mmol,2.33g)和乙酸乙酯(10mL),20℃左右搅拌30分钟,降温到内温-80℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为57%,液相检测de值为19%。
实施例3 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,2.31g)和二氯甲烷(10mL),20℃左右搅拌30分钟,降温到内温-30℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为42%,液相检测de值为61%。
实施例4 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,3.03g)和二氯甲烷(10mL),20℃左右搅拌30分钟,降温到内温-30℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为54%,液相检测de值为81%。
实施例5 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,1.54g)和甲基叔丁基醚(10mL),20℃左右搅拌30分钟,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为63%,液相检测de值为37%。
实施例6 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,3.47g)和二氯乙烷(10mL),20℃左右搅拌30分钟,降温到内温-15℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为45%,液相检测de值为77%。
实施例7 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,2.43g)和二氯甲烷(10mL),20℃左右搅拌30分钟,升温到内温30℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为51%,液相检测de值为83%。
实施例8 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,3.15g)和二氯乙烷(10mL),20℃左右搅拌30分钟,降温到内温-15℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为47%,液相检测de值为89%。
实施例9 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(0.1mmol,0.11g)和二氯乙烷(10mL),20℃左右搅拌30分钟,降温到内温-15℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为48%,液相检测de值为91%。
实施例10 Ivosidenib中间体A的合成
在干燥氮气保护的反应瓶中加入L-焦谷氨酸(0.1mol,12.91g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(0.1mmol,0.12g)和二氯乙烷(10mL),20℃左右搅拌30分钟,降温到内温5℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib中间体A,收率为53%,液相检测de值为94%。
实施例11 Ivosidenib的合成
在干燥氮气保护的反应瓶中加入N-(4-氰基吡啶-2-基)L-焦谷氨酸(0.1mol,23.12g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,2.31g)和二氯甲烷(10mL),20℃左右搅拌30分钟,降温到内温0℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib,收率为61%,液相检测de值为65%。
实施例12 Ivosidenib的合成
在干燥氮气保护的反应瓶中加入N-(4-氰基吡啶-2-基)L-焦谷氨酸(0.1mol,23.12g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,3.03g)和二氯甲烷(10mL),20℃左右搅拌30分钟,降温到内温0℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib,收率为58%,液相检测de值为71%。
实施例13 Ivosidenib的合成
在干燥氮气保护的反应瓶中加入N-(4-氰基吡啶-2-基)L-焦谷氨酸(0.1mol,23.12g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,2.43g)和二氯甲烷(10mL),20℃左右搅拌30分钟,降温到内温0℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib,收率为50%,液相检测de值为84%。
实施例14 Ivosidenib的合成
在干燥氮气保护的反应瓶中加入N-(4-氰基吡啶-2-基)L-焦谷氨酸(0.1mol,23.12g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,3.15g)和二氯甲烷(10mL),20℃左右搅拌30分钟,降温到内温0℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib,收率为56%,液相检测de值为90%。
实施例15 Ivosidenib的合成
在干燥氮气保护的反应瓶中加入N-(4-氰基吡啶-2-基)L-焦谷氨酸(0.1mol,23.12g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(3mmol,3.15g)和甲苯(10mL),20℃左右搅拌30分钟,降温到内温0℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib,收率为50%,液相检测de值为85%。
实施例16 Ivosidenib的合成
在干燥氮气保护的反应瓶中加入N-(4-氰基吡啶-2-基)L-焦谷氨酸(0.1mol,23.12g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(0.1mmol,0.11g)和二氯甲烷(10mL),20℃左右搅拌30分钟,降温到内温5℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib,收率为55%,液相检测de值为92%。
实施例17 Ivosidenib的合成
在干燥氮气保护的反应瓶中加入N-(4-氰基吡啶-2-基)L-焦谷氨酸(0.1mol,23.12g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(0.1mmol,0.12g)和二氯甲烷(10mL),20℃左右搅拌30分钟,降温到内温-20℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。反应液用5%冰的碳酸钠溶液(30mL)洗涤2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶纯化后得光学纯的Ivosidenib,收率为57%,液相检测de值为96%。
实施例18 Ivosidenib的合成
在干燥氮气保护的反应瓶中加入N-(4-氰基吡啶-2-基)L-焦谷氨酸(0.1mol,23.12g)、邻氯苯甲醛(0.1mol,14.06g)、3-氨基-5-氟吡啶(0.1mol,11.21g)、手性质子酸催化剂(0.1mmol,0.12g)和甲醇(10mL),20℃左右搅拌30分钟,降温到内温-10℃左右,缓慢滴加1,1-二氟-3-异环丁腈(0.1mol,11.71g),滴加完毕,反应过夜。旋干溶剂,加入二氯甲烷(20mL)溶解,用5%冰的碳酸钠溶液(30mL)洗涤有机相2次,无水硫酸钠干燥,旋干,经二氯甲烷/正己烷(1:3)重结晶得光学纯的Ivosidenib,收率为59%,,液相检测de值为93%。
一种Ivosidenib及其中间体的手性合成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。






![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)








动态评分
0.0