

IPC分类号 : C07D487/04,C07B59/00,A61K51/04,A61K101/02










专利摘要
本发明公开了一种以7‑脱氮腺嘌呤碱基为母核的18F‑PET/CT示踪剂及其制备方法。本发明首先公开了式Ⅰ所示化合物或其药学上可接受的盐,其中,R1、R2分别独立地选自H或C1‑C5的烷基;X独立选自11C、18F、15O、或13N。本发明还公开了式Ⅰ所示化合物的制备方法和应用。本发明成功制备得到了一种以7‑脱氮腺嘌呤碱基为母核的18F‑PET/CT示踪剂:18F‑HX示踪剂。实验结果表明,本发明制备的18F‑HX示踪剂可在肿瘤中摄取稳定,高于肌肉组织的3倍。因此,本发明18F‑PET/CT示踪剂有望应用于PET/CT影像学方面,改变在肺癌上示踪剂缺乏的局面。
权利要求
1.一种化合物或药学上可接受的盐,其特征在于:所述化合物为如下化合物
2.一种制备权利要求1所述化合物
(1)化合物1、四甘醇双(对甲苯盐酸盐)和无水碳酸钾置于有机溶液中回流加热后,HCl中和,萃取,减压干燥,色谱分离,即得化合物2;所述化合物1、四甘醇双(对甲苯盐酸盐)和无水碳酸钾的摩尔比为1:1:1;
(2)化合物2溶于DMF后置于PET单次模块中,打靶,除水,亲核取代反应后,纯化,洗脱,即得
制备路线如下:
3.根据权利要求2所述的方法,其特征在于:
步骤(1)中,所述有机溶液为苯溶液;
和/或,步骤(1)中,所述回流加热时间为4h;
和/或,步骤(1)中,所述HCl的浓度为1N;
和/或,步骤(1)中,所述萃取液为二氯甲烷;
和/或,步骤(1)中,所述色谱分离的流动相为体积比100:1~2的二氯甲烷与甲醇混合溶液;
和/或,步骤(2)中,所述PET单次模块中,乙醇活化C18柱,碳酸氢钠活化QMA柱;
和/或,步骤(2)中,所述打靶时间为30~60min;
和/或,步骤(2)中,所述除水剂为无水乙腈;
和/或,步骤(2)中,所述亲核取代反应温度为125℃,时间为12min;
和/或,步骤(2)中,所述纯化使用C18柱分离纯化;
和/或,步骤(2)中,所述洗脱使用无水乙醇。
4.根据权利要求2所述的方法,其特征在于:所述化合物1的制备方法包括如下步骤:
1)化合物3与碳酸铯溶于DMF中,混合后冷却至0℃,加入碘代正丁烷,并在室温下搅拌,发生烷基化反应,反应后混合液冷却,沉淀,抽滤,干燥后即得化合物4;所述化合物3、碳酸铯与碘代正丁烷的摩尔比为1:0.2~0.3:2~4;
2)化合物4、4-羟基苯硼酸、四(三苯基膦)钯和碳酸钾溶于1,4-二氧六环和水混合溶液反应,反应完后减压蒸出溶剂,溶于水和乙酸乙酯的混合溶液中,合并有机相,干燥,减压旋蒸溶剂,分离纯化,即得化合物5;所述化合物4、4-羟基苯硼酸、四(三苯基膦)钯和碳酸钾的摩尔比为1:2:0.015~0.017:1~2;
3)在高压反应釜中加入化合物5,并加入1,4-二氧六环溶解后,加入氨水反应,萃取,干燥,抽滤,减压蒸发溶剂,分离纯化,即得化合物1;
制备路线如下:
5.根据权利要求4所述的方法,其特征在于:和/或,步骤1)中,所述搅拌时间为5h;
和/或,步骤1)中,所述化合物3、碳酸铯与碘代正丁烷的摩尔比为1:0.2~0.3:3;
和/或,步骤2)中,所述1,4-二氧六环和水混合后的溶液中1,4-二氧六环和水的体积比为4:1;
和/或,步骤2)中,所述反应通入氮气保护,反应温度为95℃;
和/或,步骤2)中,所述水和乙酸乙酯的混合溶液中水和乙酸乙酯的体积比为4:6;
和/或,步骤2)和步骤3)中,所述干燥使用无水硫酸钠;
和/或,步骤2)和步骤3)中,所述分离纯化使用柱层析法;
和/或,步骤3)中,所述氨水浓度为28%;
和/或,步骤3)中,高压反应釜温度为120℃,反应时间为24h。
6.权利要求1所述的化合物在制备示踪剂中的应用;其中,所述示踪剂应用于医学影像诊断领域。
7.根据权利要求6所述的应用,其特征在于:所述示踪剂为PET/CT示踪剂。
8.根据权利要求7所述的应用,其特征在于:所述PET/CT示踪剂在肿瘤诊断中的应用。
9.根据权利要求8所述的应用,其特征在于:所述肿瘤为肺癌。
10.一种示踪剂,它是以权利要求1所述的化合物或其药学上可接受的盐,加上药学上可接受的辅料制备而成的。
说明书
技术领域
本发明属于有机化学领域,具有涉及一种以7-脱氮腺嘌呤碱基为母核的18F-PET/CT示踪剂及其制备方法。
背景技术
PET/CT作为一种功能显像的先进诊疗设备,虽然其在肺癌中的应用日益得到推广,但是仍然有极大的局限性。这种局限性主要来源于功能影像的核心成分:PET/CT示踪剂。当前肺癌的PET/CT示踪剂主要为18F-FDG,由于18F-FDG缺乏除糖代谢之外其它分子层面的特异性,一方面存在假阳性与假阴性的缺陷,另一方面不能在功能影像层面反映肺癌的分子分型或其他特异分子靶标的信息,成为肺癌精准诊治的重大技术瓶颈。基于上述现状,目前各国研究与医疗机构开展了新型PET/CT示踪剂的基础与临床探索,这方面的研发已经成为功能影像领域的热点。例如,目前已有针对DNA合成水平增高的18F-FLT(TK1为靶标)和针对核苷代谢紊乱的18F-ACT(dCK为靶标)的PET示踪剂。临床研究表明,18F-FLT和18F-FDG的联用在一定程度上弥补了单一显像的不足,能够帮助临床医生做出更加准确的诊断和鉴别,并进一步指导临床分期和制定个体化治疗方案。此外,也有其它肿瘤的新型示踪剂的报道,例如18F-甲基胆碱在膀胱癌的鉴别诊断方面明显优于18F-FDG。但是,目前能用于肺癌诊断的示踪剂种类还非常有限,因此亟需开发用于肺癌精准诊断的新型功能影像示踪剂和前体试剂。
发明内容
本发明的目的在于提供一种以7-脱氮腺嘌呤碱基为母核的18F-PET/CT示踪剂及其制备方法。
本发明提供了式Ⅰ所示化合物或其药学上可接受的盐:
其中,R1、R2分别独立地选自H或C1-C5的烷基;
X独立选自11C、18F、15O、或13N。
进一步地,所述化合物为如下化合物式Ⅱ:
其中,X独立选自11C、18F、15O、或13N。
进一步地,所述化合物为如下化合物18F-HX:
本发明还提供了一种制备前述化合物18F-HX的方法,它包括以下步骤:
(1)化合物1、四甘醇双(对甲苯盐酸盐)和无水碳酸钾置于有机溶液中回流加热后,HCl中和,萃取,减压干燥,色谱分离,即得化合物2;所述化合物1、四甘醇双(对甲苯盐酸盐)和无水碳酸钾的摩尔比为1:1:1;
(2)化合物2溶于DMF后置于PET单次模块中,打靶,除水,亲核取代反应后,纯化,洗脱,即得18F-HX;
制备路线如下:
优选地,步骤(1)中,所述有机溶液为苯溶液;
和/或,步骤(1)中,所述回流加热时间为4h;
和/或,步骤(1)中,所述HCl的浓度为1N;
和/或,步骤(1)中,所述萃取液为二氯甲烷;
和/或,步骤(1)中,所述色谱分离的的流动相为体积比100:1~2的二氯甲烷与甲醇混合溶液;
和/或,步骤(2)中,所述PET单次模块中,乙醇活化C18柱,碳酸氢钠活化QMA柱;
和/或,步骤(2)中,所述打靶时间为30~60min;
和/或,步骤(2)中,所述除水剂为无水乙腈;
和/或,步骤(2)中,所述亲核取代反应温度为125℃,时间为12min;
和/或,步骤(2)中,所述纯化使用C18柱分离纯化;
和/或,步骤(2)中,所述洗脱使用无水乙醇。
进一步地,所述化合物1的制备方法包括如下步骤:
1)化合物3与碳酸铯溶于DMF中,混合后冷却至0℃,加入正丁基,并在室温下搅拌,发生烷基化反应,反应后混合液冷却,沉淀,抽滤,干燥后即得化合物4;所述化合物3、碳酸铯与正丁基的摩尔比为1:0.2~0.3:2~4;
2)化合物4、4-羟基苯硼酸、四(三苯基膦)钯和碳酸钾溶于1,4-二氧六环和水混合溶液反应,反应完后减压蒸出溶剂,溶剂溶于水和乙酸乙酯的混合溶液中,合并有机相,干燥,减压旋蒸溶剂,分离纯化,即得化合物5;所述化合物4、4-羟基苯硼酸、四(三苯基膦)钯和碳酸钾的摩尔比为1:2:0.015~0.017:1~2;
3)在高压反应釜中加入化合物5,并加入1,4-二氧六环溶解后,加入氨水反应,萃取,干燥,抽滤,减压蒸发溶剂,分离纯化,即得化合物1;
制备路线如下:
优选地,步骤1)中,所述正丁基为碘代正丁烷;
和/或,步骤1)中,所述搅拌时间为5h;
和/或,步骤1)中,所述化合物3、碳酸铯与正丁基的摩尔比为1:0.2~0.3:3;
和/或,步骤2)中,所述1,4-二氧六环和水混合后的溶液中1,4-二氧六环和水的体积比为4:1;
和/或,步骤2)中,所述反应通入氮气保护,反应温度为95℃;
和/或,步骤2)中,所述水和乙酸乙酯的混合溶液中水和乙酸乙酯的的体积比为4:6;
和/或,步骤2)和步骤3)中,所述干燥使用无水硫酸钠;
和/或,步骤2)和步骤3)中,所述分离纯化使用柱层析法;
和/或,步骤3)中,所述氨水浓度为28%;
和/或,步骤3)中,高压反应釜温度为120℃,反应时间为24h。
本发明还提供了前述的化合物在制备示踪剂中的应用;其中,所述示踪剂应用于医学影像诊断领域。
进一步地,所述示踪剂为PET/CT示踪剂。
进一步地,所述PET/CT示踪剂在肿瘤诊断中的应用。
进一步地,所述肿瘤为肺癌。
本发明还提供了一种示踪剂,它是以前述化合物或其药学上可接受的盐,加上药学上可接受的辅料或者辅助性成分制备而成的。
本发明成功制备得到了一种以7-脱氮腺嘌呤碱基为母核的18F-PET/CT示踪剂:18F-HX示踪剂。实验结果表明,本发明制备的18F-HX示踪剂可在肿瘤中摄取稳定,高于肌肉组织的3倍。因此,本发明18F-PET/CT示踪剂有望应用于PET/CT影像学方面,改变在肺癌上示踪剂缺乏的局面。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
图1为化合物1的放射性核素标记。
图2为18F-HX的TLC检测数据分析。
图3位18F-HX在肌肉与裸鼠肿瘤部位摄取的对比分析。
具体实施方式
实施例1化合物1的制备
(1)5-碘-7-正丁基-7H-吡咯并[2,3-d]嘧啶4-氯(化合物4)的合成:
4-氯-5-碘-7H-吡咯并[2,3-d]嘧啶(化合物3)(5g,17.90mmol)与碳酸铯(11.66g,4.66mmol)混合溶于30mL DMF并置于冰水中,待混合液冷却到0℃,加入碘代正丁烷(6.17mL,53.7mmol)。10min之后,将混合液从冰水中取出放在室温密封搅拌5h,用TLC检测反应进程。反应完成后,将混合液冷却并加入60mL冰水,出现沉淀。抽虑,干燥后得黄色固体(化合物4)5.52g(产率为91.9%)。
化合物4:1H NMR(600MHz,DMSO-d6)δ8.64(s,1H),8.06(s,1H),4.27(t,J=7.1Hz,2H),1.82–1.74(m,2H),1.22(h,J=7.4Hz,2H),0.88(t,J=7.4Hz,3H).13C NMR(151MHz,DMSO-d6)δ162.75,151.34,150.87,150.75,136.89,116.47,51.59,44.89,36.25,31.89,31.24,19.71,13.84.HRMS(ESI-TOF):for C10H12ClIN3[M+H]:calcd 335.9765;found 335.9773.
(2)4-(4-氯-7-正丁基-7H-吡咯并[2,3-d]嘧啶-5-)苯酚(化合物5)的合成:
将化合物4(1.0g,2.99mmol),4-羟基苯硼酸(826mg,5.98mmol),四(三苯基膦)钯(173mg,0.05mmol)以及碳酸钾(622mg,4.49mmol)混合后溶于1,4-二氧六环/水(4:1,50mL),并充入氮气保护。反应混合液加热到95℃,用TLC监测反应。反应完成后,减压蒸出溶剂,再溶于水/乙酸乙酯(4:6)50mL×3次。将有机相合并,用无水硫酸钠干燥后,减压旋蒸溶剂。通过柱层析分离纯化得到白色固体(化合物5)0.51g(产率为70.7%)。
化合物5:1H NMR(400MHz,DMSO-d6)δ9.53(s,1H),8.15(s,1H),7.33–7.20(m,3H),6.93–6.80(m,2H),4.17(t,J=7.1Hz,2H),3.37(s,1H),1.79(p,J=7.2Hz,2H),1.37–1.22(m,3H),0.92(t,J=7.3Hz,3H).13C NMR(101MHz,DMSO-d6)δ157.17,156.37,151.33,149.90,129.63,129.33,125.29,122.44,115.69,115.07,115.02,100.03,43.25,31.80,31.66,28.95,19.36,13.45.HRMS(ESI-TOF):for C16H17ClN3O[M+H]:calcd 302.1061;found 302.1060.
(3)4-(4-氨基-7-正丁基-7H-吡咯并[2,3-d]嘧啶-5-)苯酚(化合物1)的合成:
在高压反应釜中加入化合物5(130mg,0.3mmol),加入3mL 1,4-二氧六环溶解后,再加入28%的氨水3mL。高压釜调温到120℃,反应24h。反应液浓缩后,用二氯甲烷/水萃取。有机层用无水硫酸钠干燥后抽虑并减压蒸发溶剂。粗产物用柱层析分离纯化得到白色固体(化合物1)92.83mg(产率为76.2%)。
化合物1:1HNMR(600MHz,Chloroform-d)δ8.25(s,1H),7.55-7.05(d,J=7.4Hz,4H),7.01(s,1H),6.99(s,1H),5.36(s,2H),4.26-4.23(t,J=7.2Hz,2H),1.89–1.83(m,2H),1.41–1.36(m,2H),0.89-0.87(d,3H).HR-MS(ESI+):Calc.for[C16H18N4O]:283.1514[M+H]+;Found 283.1552[M+H]+,305.1386[M+Na]+.
实施例2 18F-HX化合物的制备
1、化合物2的制备
四甘醇双(对甲苯盐酸盐)(164mg,3.46mmol),化合物1(100mg,3.46mmol)和无水碳酸钾(100mg,3.46mol)于50mL苯溶液中回流加热4h,1N HCl的中和后,将反应混合物用二氯甲烷萃取。然后将溶液减压干燥,得到固体后进行柱色谱分离得到棕黄色糖浆状产物(化合物2)160mg(产率为76%)。
化合物2:1H NMR(600MHz,Chloroform-d)δ8.29(s,1H),δ7.84-6.97(m,8H,arom-H),6.95(s,1H),5.52(s,2H),4.23-4.21(t,J=7.2Hz,2H),4.23-3.59(t,J=7.2Hz,16H),δ2.44(s,3H),1.87-1.80(m,2H),1.44-1.35(m,2H),0.97-0.95(t,J=7.4Hz,3H).HR-MS(ESI+):Calc.for[C31H40N4O7S]:613.2618[M+H]+;Found 613.2694[M+H]+,635.2521[M+Na]+.
2、18F-HX的制备
化合物1的的放射性核素标记如图1所示。18F-HX的制备过程如下所示:
(1)取1mg化合物2,溶于0.8mL DMF中;
(2)PET单次模块准备:
a.取0.5M碳酸氢钠10mL冲洗QMA柱,再用10mL高纯水冲洗,活化QMA柱;
b.取5mL乙醇冲洗C-18柱,再用10mL高纯水冲洗,对C-18柱进行纯化;
c.设置如下程序:
B1:K22乙腈淋洗液1.5mL淋洗QMA柱,洗脱QMA柱子上的18F+到反应管;
B2:在反应管中加入无水乙腈2mL,进行除水;
B3:反应管冷却后加入0.8mL化合物2,在125℃下进行亲核反应,反应时间为12min;
B4:加水将产物转移至C-18柱分离纯化,继续用水清洗,除去QMA柱上残留的18F+离子以及乙腈等杂质;
B5:用5mL无水乙醇洗脱18F-HX。
(3)制备及分析:
A.打靶30-60min,18F离子约600mCi转入合成模块;
B.按照(2)中设置的程序B1~B5进行18F-HX的合成;
C产物分离后,C-18柱监测到约300-360mCi残留,为未标记的18F离子;
D.终产物放射性活度检测约10mCi,体积约5mL;
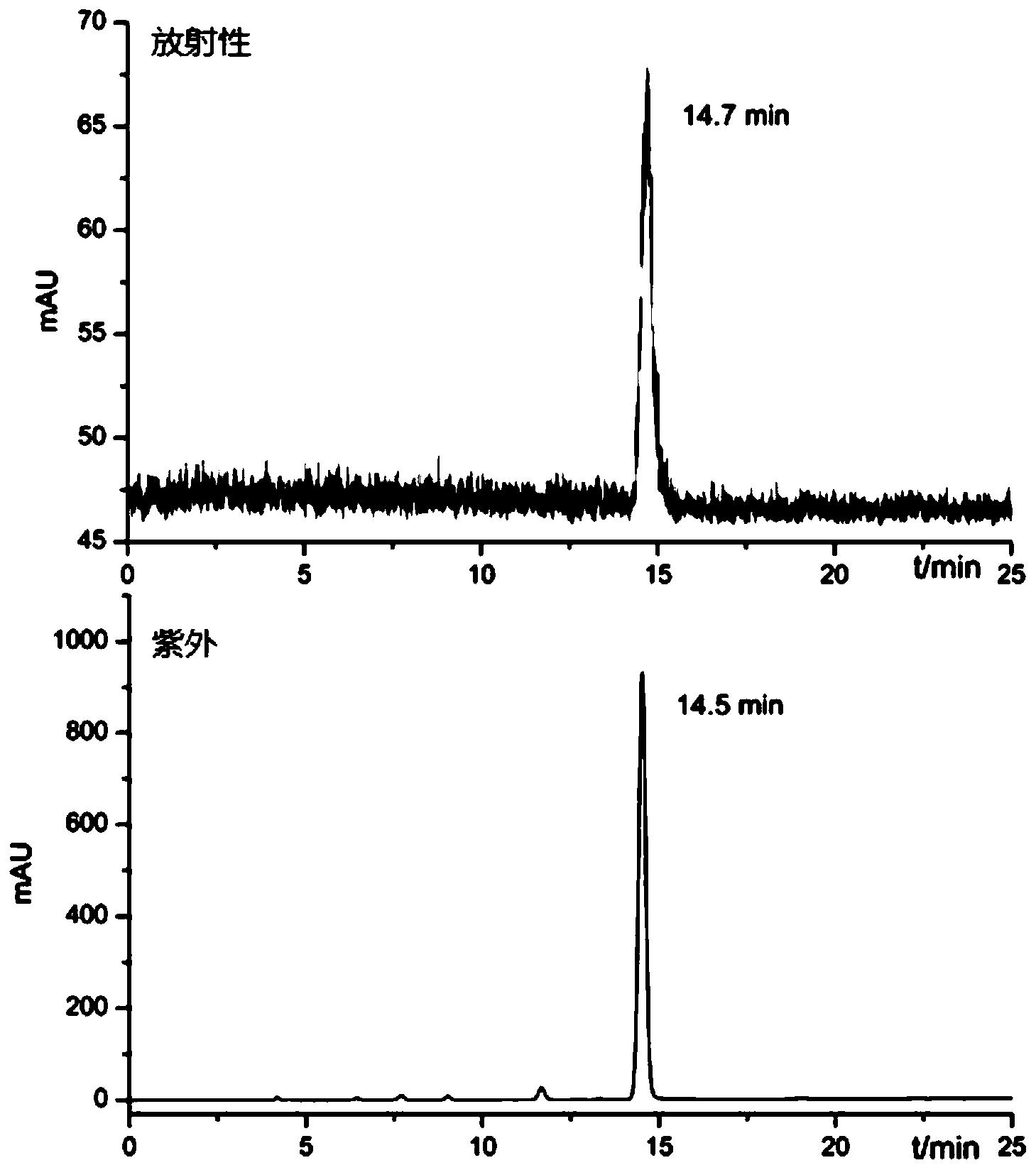
E.85%乙腈流动相,TLC检测,分析F离子峰与产物峰的占比(图2)。
将薄层板下沿浸泡于流动相,点板后,经过放射性检测器检测,可得到含放射性标记的核素探针的峰位置及峰度。根据图2可知,游离F离子Rf=0.263,18F-HX的Rf=0.593,产物峰度约为88.93%。在整个过程中18F的标记率约为15.9%,经过HPLC对标记的化合物进行纯化与鉴定后,得到放化纯度大于95%的18F-HX化合物。
以下通过试验例的方式来说明本发明的有益效果。
本发明的试验例中的荷瘤小鼠是接种了肺腺癌上皮细胞A549而肿瘤模型制造成功的小鼠。
试验例1 18F-HX示踪剂在荷瘤小鼠体内成像的研究
1、实验材料:
荷瘤小鼠不少于12只,18F-HX,18F-FDG,吸水纸,棉签,酒精棉球,胰岛素针,取材手术器械一套,称量纸、放免管、万分之一天平、生理盐水。
2、实验步骤
荷瘤小鼠随机分为4组,每组N≥3,提前禁食6h以上,可喝水。
放免管提前称重并记录空管重量。
尾静脉注射18F-HX试剂37kBq(每只10μCi)。
分别在注射后10min,30min,60min将小鼠处死,并取血液,心,肝,肺,脾,胃,肠,肾,脑,肌肉,骨,肿瘤组织,置于放免管中,称重并记录管重量,计算出组织重量。
注射后准备三管标准品,将含有组织的放免管放入Gamma-counter中,记录放射性数值,根据放射性数值/组织重量,计算出每g不同组织中的放射性剂量分布ID%/g,计算过程如下:
第一步,计算标准样品的CPM值,求得平均值Vstd(每个数减去背景值,再求平均),计算出注射剂量的1%。即为Vstd%。
第二步,每一个样品的CPM计数后,通过衰减校正还原后,得到校正后的CPM。利用这个值除以Vstd%,得到ID%。
第三步,利用得到的ID%除以重量g,得到ID%/g。把数据制成图像。
2SD%表示的95%可置信区间,在估计总体参数时,一般都会给出一个较高的置信度,如95%或99%等。但是,当样本容量n为一定时,置信度越高,置信区间就越大,也即估计的参数的相对精度就会越低。反之,置信度越低,则精度相对就会越高。
给荷瘤小鼠尾静脉注射18F-FDG作为对照组。
3、实验结果
通过对注射18F-HX示踪剂后的10min,30min,60min体内分布进行研究,各个脏器的扫描对比结果表明,18F-HX主要聚集在肝脏,肾脏中。并且随着时间的增加,肿瘤组织的摄取量比较稳定,而肌肉的摄取量在逐渐降低。从30min及60min的扫描结果可以看出,肿瘤组织对18F-HX具有很高的摄取,已经超过肌肉组织的3倍,如图3。
综上,本发明成功制备得到了一种以7-脱氮腺嘌呤碱基为母核的18F-PET/CT示踪剂:18F-HX示踪剂。本发明制备的18F-HX示踪剂可在肿瘤中摄取稳定,高于肌肉组织的3倍。因此,本发明18F-PET/CT示踪剂有望应用于PET/CT影像学方面,改变在肺癌上示踪剂缺乏的局面。
一种以7-脱氮腺嘌呤碱基为母核的18F-PET/CT示踪剂及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。







![酞嗪并[1,2,b]喹唑啉-8-酮化合物及其制备方法和在抗肿瘤药物中的应用](https://www.zhichawang.com/images/tongyong/wutu.jpg)






![一种吲哚[3,2-b]咔唑为核的空穴传输材料及其制备方法和应用](https://www.zhichawang.com/images/CN111171036A/CN111171036A.jpg)





动态评分
0.0