

IPC分类号 : C07J43/00,A61K51/04,A61K31/58,A61P35/04,A61P35/00,A61P15/14,A61K101/02




专利摘要
本发明涉及药物化学领域,具体涉及一种18F标记的炔雌醇及其制备方法和应用,在炔雌醇上引入修饰基团并经过18F标记,具备良好的生物活性和优异的药动学性质,而且体外稳定性好,对乳腺癌ER+细胞具有较好的靶向性,可以作为PET示踪剂,用于高转移乳腺癌ER+细胞的早期诊断或疗效评价。本发明还提供18F标记炔雌醇的制备方法,以炔雌醇为原料,通过Click反应引入标记基团,在相对温和的条件下加热,通过18F‑交换,简化标记步骤和纯化时间,所获得的18F标记的炔雌醇放射化学产率高,更加有利于放射性标记化合物的商业应用于临床推广。
权利要求
1.一种18F标记的炔雌醇,其特征在于,具有式(I)结构,
其中,R1选自烷基或烷氧基。
2.根据权利要求1所述的18F标记的炔雌醇,其特征在于,所述R1为亚乙基或3,6,9,12-四氧杂十四烷基。
3.一种制备18F标记炔雌醇的方法,其特征在于,所述方法包括如下步骤:
(1)取炔雌醇溶解于与水互溶性较好有机溶剂中,将式A-I所示的化合物加入上述的炔雌醇溶液中,然后加入CuSO4·5H2O的水溶液、NaAsC的水溶液和三(2-苯并咪唑基甲基)胺的水溶液,于40-50℃下反应0.5-2h,将所得反应液旋干,加入与水互溶性较好有机溶剂并离心,取上清液,即得式A-II所示的化合物;
(2)将所述式A-II所示的化合物溶于N,N-二甲基甲酰胺中,然后加入18F-溶液中,于80-100℃下反应10-40min,将所得反应液进行纯化、洗涤,即得所述式(I)所示的化合物;
4.根据权利要求3所述的方法,其特征在于,所述步骤(1)中,炔雌醇、式A-I所示的化合物、CuSO4·5H2O、抗坏血酸钠和三(2-苯并咪唑基甲基)胺加入的摩尔比为1∶(0.90~1.30)∶(0.18~0.22)∶(1.8~2.2)∶(0.08~0.12)。
5.根据权利要求3或4所述的方法,其特征在于,在所述步骤(1)反应体系中,所述与水互溶性较好有机溶剂与水的体积比为1∶1。
6.根据权利要求3-5任一项所述的方法,其特征在于,式A-I所示的化合物如下,
上述式A-I-1所示的化合物的合成步骤包括:
Q1、将式X-1所示的化合物2-(二甲氨基)溴乙烷氢溴酸盐与叠氮化钠混合,加水溶解,氮气保护,87~93℃下反应15~17h,将所得反应液调节pH值至中性,将上述反应液进行TLC检测,以二氯甲烷∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.5,采用二氯甲烷萃取,所得有机相经干燥,旋干,得油状液体,即为式X-2所示的化合物;
Q2、将式X-2所示的化合物加入醚类溶剂中溶解,氮气保护,然后逐滴加入溴甲基硼酸频哪醇酯的醚类溶液,滴加完毕,20-30℃继续反应2~4h,至出现白色浑浊,用醚类溶剂沉淀,离心,真空干燥,得到化合物X-3;
Q3、向式X-3所示的化合物中加入氟氢化钾、盐酸、水、与水互溶性较好的有机溶剂,42~47℃下反应1~3h,将上述反应液进行TLC检测,以乙酸乙酯∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.3,用乙酸乙酯萃取,旋干,纯化,得到上述式A-I-1所示的化合物;
7.根据权利要求3-6任一项所述的方法,其特征在于,式A-I所示的化合物如下,
上述式A-I-2所示的化合物的合成步骤包括:
F1、将式Y-1所示的四聚乙二醇溶解于极性非质子性溶剂中,加入碱液,缓慢滴加含对甲苯磺酰氯的溶液,滴加完毕,置于冰浴中反应1.5~2.5h,萃取,纯化,得到油状物,即式Y-2所示的化合物;
F2、将式Y-2所示的化合物与叠氮化钠溶于极性非质子性溶剂中,93~97℃下加热反应15~17h,萃取,旋蒸,得到油状物,即式Y-3所示的化合物;
F3、将式Y-3所示的化合物溶于极性非质子性溶剂中,加入4-二甲氨基吡啶和三乙胺,冰浴下搅拌,缓慢滴加含对甲苯磺酰氯的溶液,滴加完毕,室温反应过夜,然后经洗涤、旋蒸和纯化,得到黄色油状物,即式Y-4所示的化合物;
F4、将式Y-4所示的化合物、N,N-二甲基乙醇胺、碱溶于极性非质子性溶剂中,67~72℃加热反应10~12h,过滤,收集滤液,经旋蒸、萃取、旋干和纯化,得式Y-5所示的化合物;
F5、将式Y-5所示的化合物溶于干燥的极性非质子性溶剂中,将溴甲基硼酸频那醇酯溶于干燥的的极性非质子性溶剂中,然后将上述溴甲基硼酸频那醇酯的溶液逐滴加入上述式Y-5所示的化合物的溶液中,在室温、氮气保护的条件下反应2.5~3.5h,旋干,得到式Y-6所示的化合物;
F6、将式Y-6所示的化合物溶于与水互溶性较好的有机溶剂中,加入氟氢化钾水溶液、盐酸,氮气保护,42~47℃反应1.5~2.5h,加入氨水淬灭,调节pH值至中性,旋干,纯化,得到上述式A-I-2所示的化合物;
8.权利要求1或2所述的18F标记的炔雌醇在制备治疗乳腺癌药物中的应用。
9.权利要求1或2所述的18F标记的炔雌醇在制备PET示踪剂中的应用。
10.一种PET示踪剂,其特征在于,包括权利要求1或2所述的18F标记的炔雌醇。
说明书
技术领域
本发明涉及药物化学领域,具体涉及一种18F标记的炔雌醇及其制备方法和应用。
背景技术
乳腺癌是女性易患的常见癌症,易发生转移,发病率逐年上升且呈年轻化,严重影响女性健康。临床研究发现,雌激素受体阳性乳腺癌的浸润性行为较低,且对内分泌治疗较为敏感。但经过一阶段内分泌治疗后,病灶的雌激素受体活性可发生改变进而影响后续疗效。免疫病理检查只能获取手术病灶的雌激素受体分布情况,而原发灶受体的情况并不能完全代表转移灶,尤其是治疗病灶雌激素受体的活性。因此,在早期正确的判断乳腺癌治疗过程中各部分病灶的雌激素受体活性状况,是及时调整治疗方案和建立个体化治疗的关键。
正电子发射型计算机断层显像(Positron Emission Computed Tomography,简称PET),是核医学领域比较先进的临床检查影像技术。目前,PET在肿瘤、冠心病和脑部疾病这三大类疾病的诊疗中尤其显示出重要的价值。16α-18F-17β-雌二醇(16α-[18F]-fluoro-17β-estradiol,18F-FES)是一种雌二醇衍生物,能特异性地与雌激素受体结合,作为雌激素受体的分子影像探针,通过PET/CT显像能动态、定量和无创伤地反映体内雌激素受体表达水平和分布情况,为乳腺癌患者提供一种有力评价方法。乳腺癌患者的18F-FES PET和PET/CT显像的临床实验结果发现,乳腺癌对18F-FES的吸收与雌激素受体的表达水平有明显的相关性,而且,抗雌激素治疗后18F-FES的吸收明显减少。随着18F-FES PET和PET/CT在女性乳腺癌领域的应用进一步扩展,18F-FES的合成已成为亟需解决的问题。
1984年,Dale O.Kiesewetter等人报道了在16α-17β-雌二醇的五元环上直接引入[18F]氟离子进行标记实验,但是该方法标记条件苛刻,需要在-78℃下加入氢化铝锂进行反应,这给标记实验增加了很大的难度和危险性,不具有通用性。1995年,Jojannes Romer等人改善了标记条件,首先将16α-17β-雌二醇引入磺酰基,然后在磺酰基上引入[18F]氟离子进行标记,虽然反应条件相对温和,但是该标记反应需要两步才能完成,时间较长,放射化学产率较低。如何对现有的雌二醇衍生物的合成路线进行改进并标记以最大限度地利用PET技术清楚判断雌激素受体活性情况,为患者及时调整治疗方案,建立个体化治疗,已成为本领域亟待解决的一大技术难题。
发明内容
本发明要解决的技术问题在于克服现有技术中18F标记的炔雌醇合成和标记反应条件苛刻且在用药过程存在严重不良反应的缺陷,从而提供一种疗效安全、易于合成和标记,并能利用PET技术以清楚判断雌激素受体活性情况的18F标记的炔雌醇。
为此,本发明提供了一种18F标记的炔雌醇,具有式(I)所示的结构,
其中,R1选自烷基或烷氧基。
优选的,R1为亚乙基或3,6,9,12-四氧杂十四烷基。
本发明提供了一种制备18F标记炔雌醇的方法,包括如下步骤:
(1)取炔雌醇溶解于与水互溶性较好有机溶剂中,将式A-I所示的化合物加入上述的炔雌醇溶液中,然后加入CuSO4·5H2O的水溶液、NaAsC的水溶液和三(2-苯并咪唑基甲基)胺的水溶液,于40-50℃下反应0.5-2h,将所得反应液旋干,加入与水互溶性较好有机溶剂并离心,取上清液,即得式A-II所示的化合物;
(2)将所述式A-II所示的化合物溶于N,N-二甲基甲酰胺中,然后加入18F-溶液中,于80-100℃下反应10-40min,将所得反应液进行纯化、洗涤,即得所述式(I)所示的化合物;
优选的,所述步骤(1)中,炔雌醇、式A-I所示的化合物、CuSO4·5H2O、抗坏血酸钠和三(2-苯并咪唑基甲基)胺加入的摩尔比为1∶(0.90~1.30)∶(0.18~0.22)∶(1.8~2.2)∶(0.08~0.12)。
优选的,在所述步骤(1)反应体系中,所述与水互溶性较好有机溶剂与水的体积比为1∶1。
优选的,式A-I所示的化合物如下,
上述式A-I-1所示的化合物的合成步骤包括:
Q1、将式X-1所示的化合物2-(二甲氨基)溴乙烷氢溴酸盐与叠氮化钠混合,加水溶解,氮气保护,87~93℃下反应15~17h,将所得反应液调节pH值至中性,将上述反应液进行TLC检测,以二氯甲烷∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.5,采用二氯甲烷萃取,所得有机相经干燥,旋干,得油状液体,即为式X-2所示的化合物;
Q2、将式X-2所示的化合物加入醚类溶剂中溶解,氮气保护,然后逐滴加入溴甲基硼酸频哪醇酯的醚类溶液,滴加完毕,20-30℃继续反应2~4h,至出现白色浑浊,用醚类溶剂沉淀,离心,真空干燥,得到化合物X-3;
Q3、向式X-3所示的化合物中加入氟氢化钾、盐酸、水、与水互溶性较好的有机溶剂,42~47℃下反应1~3h,将上述反应液进行TLC检测,以乙酸乙酯∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.3,用乙酸乙酯萃取,旋干,纯化,得到上述式A-I-1所示的化合物;
优选的,式A-I所示的化合物如下,
上述式A-I-2所示的化合物的合成步骤包括:
F1、将式Y-1所示的四聚乙二醇溶解于极性非质子性溶剂中,加入碱液,缓慢滴加含对甲苯磺酰氯的溶液,滴加完毕,置于冰浴中反应1.5~2.5h,萃取,纯化,得到油状物,即式Y-2所示的化合物;
F2、将式Y-2所示的化合物与叠氮化钠溶于极性非质子性溶剂中,93~97℃下加热反应15~17h,萃取,旋蒸,得到油状物,即式Y-3所示的化合物;
F3、将式Y-3所示的化合物溶于极性非质子性溶剂中,加入4-二甲氨基吡啶和三乙胺,冰浴下搅拌,缓慢滴加含对甲苯磺酰氯的溶液,滴加完毕,室温反应过夜,然后经洗涤、旋蒸和纯化,得到黄色油状物,即式Y-4所示的化合物;
F4、将式Y-4所示的化合物、N,N-二甲基乙醇胺、碱溶于极性非质子性溶剂中,67~72℃加热反应10~12h,过滤,收集滤液,经旋蒸、萃取、旋干和纯化,得式Y-5所示的化合物;
F5、将式Y-5所示的化合物溶于干燥的极性非质子性溶剂中,将溴甲基硼酸频那醇酯溶于干燥的的极性非质子性溶剂中,然后将上述溴甲基硼酸频那醇酯的溶液逐滴加入上述式Y-5所示的化合物的溶液中,在室温、氮气保护的条件下反应2.5~3.5h,旋干,得到式Y-6所示的化合物;
F6、将式Y-6所示的化合物溶于与水互溶性较好的有机溶剂中,加入氟氢化钾水溶液、盐酸,氮气保护,42~47℃反应1.5~2.5h,加入氨水淬灭,调节pH值至中性,旋干,纯化,得到上述式A-I-2所示的化合物;
优选的,所述步骤(1)、Q3、F6中,所述与水互溶性好的有机溶剂包括N,N-二甲基甲酰胺、二甲亚砜、丙酮、乙腈、甲醇和乙醇中的至少一种。
优选的,所述步骤F1中,所述极性非质子性溶剂包括四氢呋喃、N,N-二甲基甲酰胺和二氧六环中的至少一种。
优选的,所述步骤F2中,所述极性非质子性溶剂包括乙腈、二甲亚砜和N,N-二甲基甲酰胺中的至少一种。
优选的,所述步骤F3中,所述极性非质子性溶剂包括二氯甲烷、四氢呋喃、N,N-二甲基甲酰胺和二甲亚砜中至少一种。
优选的,所述步骤F4、F5中,所述极性非质子性溶剂包括四氢呋喃、N,N-二甲基甲酰胺、二甲亚砜和乙腈中的至少一种。
优选的,所述的碱液包括氢氧化钾或氢氧化钠。
优选的,所述的醚类溶剂包括乙醚。
本发明提供上述18F标记的炔雌醇在制备治疗乳腺癌药物中的应用。
本发明提供上述18F标记的炔雌醇在制备PET示踪剂中的应用。
本发明还提供一种PET示踪剂,包括上述18F标记的炔雌醇。
本发明技术方案,具有如下优点:
1.本发明在炔雌醇上引入修饰基团并经过18F标记,所获得的18F标记的炔雌醇,具备良好的生物活性和优异的药动学性质,而且体外稳定性好,对乳腺癌ER+细胞具有较好的靶向性,可以作为PET示踪剂,用于高转移乳腺癌ER+细胞的早期诊断或疗效评价。
2.本发明还提供18F标记炔雌醇的制备方法,以炔雌醇为原料,通过Click反应引入标记基团,在相对温和的条件下加热,通过18F-交换,简化标记步骤和纯化时间,所获得的18F标记的炔雌醇放射化学产率高,更加有利于放射性标记化合物的商业应用于临床推广。
3.本发明还提供标记基团的制备方法,该制备方法反应速率快、生产效率高,条件简单,易于工业化控制,综合经济效益显著,解决了现有生产工艺中反应时间长、难以控制反应条件、无法满足工业化生产需求的问题。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
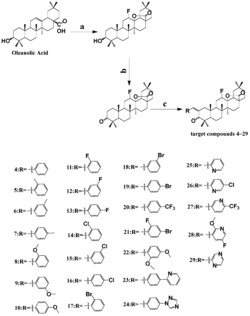
图1是本发明实施例1-6制备的18F标记的炔雌醇结构式;
图2是本发明实施例1制备的标记前体A-II-a的1H-NMR图谱;
图3是本发明实施例4制备的标记前体A-II-b的1H-NMR图谱;
图4是本发明实施例1中标记前体A-II-a的HPLC图谱;
图5是本发明实施例4中标记前体A-II-b的HPLC图谱;
图6是本发明实施例1中标记产物I-a的放射性高效液相图谱;
图7是本发明实施例4中标记产物I-b的放射性高效液相图谱;
图8是本发明实施例1中标记前体A-II-a不同温度下的标记率;
图9是本发明实施例4中标记前体A-II-b不同温度下的标记率;
图10是本发明实施例1中标记前体A-II-a体外稳定性测试图谱;
图11是本发明实施例4中标记前体A-II-b体外稳定性测试图谱;
图12是本发明实施例1中标记产物I-a在PBS中孵育4h后radio-HPLC分析图谱;
图13是本发明实施例1中标记产物I-a在血清中孵育4h后radio-HPLC分析图谱;
图14是本发明实施例1中标记产物I-a在PBS和血清中孵育不同时间的放化纯图谱;
图15是本发明实施例4中标记产物I-b在PBS中孵育4h后radio-HPLC分析图谱;
图16是本发明实施例4中标记产物I-b在血清中孵育4h后radio-HPLC分析图谱;
图17是本发明实施例4中标记产物I-b在PBS和血清中孵育不同时间的放化纯图谱;
图18是在本发明实施例1中不同浓度标记前体A-II-a下T47D细胞生存率;
图19是在本发明实施例4中不同浓度标记前体A-II-b下T47D细胞生存率;
图20是本发明实施例1中标记产物I-a在T47D细胞中的总摄取率与阻断组摄取率;
图21是本发明实施例4中标记产物I-b在T47D细胞中的总摄取率与阻断组摄取率。
具体实施方式
下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。此外,下面所描述的本发明不同实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互结合。
下述实施例中涉及到的试剂如下:
2-(二甲氨基)溴乙烷氢溴酸盐、叠氮化钠、溴甲基硼酸频哪醇酯、氟氢化钾、四聚乙二醇、对甲苯磺酰氯、N,N-二甲基乙醇胺、乙醚、乙酸乙酯、N,N-二甲基甲酰胺、CuSO4·5H2O、NaAsC、三(2-苯并咪唑基甲基)胺、四氢呋喃、二氯甲烷、甲醇、乙醇、正己烷、盐酸、氯化钠。所有试剂均为分析纯,使用前未经纯化,购置于阿拉丁试剂公司。
炔雌醇的生产厂家为安耐吉化学。
下述实施例中涉及到的仪器如下:
核磁共振仪(Bruker DRX-500)、高效液相色谱仪(waters 2445),γ计数仪(Perkin-Elmer 1470)。
实施例1
本实施例所述的具有式(I)结构18F标记的炔雌醇,如图1所示,其中R1为亚乙基,具有式(I-a)结构:
化合物(I-a)的合成路线为:
上述式I-a所示的化合物的合成步骤包括:
Q1、将式X-1所示的化合物2-(二甲氨基)溴乙烷氢溴酸盐2.33g(10mmol)与叠氮化钠2.60g(40mmol)混合,加水50ml溶解,氮气保护,90℃下反应16h,反应结束加入Na2CO3将所得反应液调节pH值至中性,将上述反应液进行TLC检测,以二氯甲烷∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.5,采用二氯甲烷萃取,所得有机相经无水硫酸钠干燥,旋干,得油状液体,质谱检测:115为M+H+,即为式X-2所示的化合物;
Q2、将式X-2所示的化合物757mg(6.63mmol)加入10ml乙醚溶解,氮气保护,然后逐滴加入含有溴甲基硼酸频哪醇酯1537.8mg(6.96mmol)的乙醚溶液5ml,滴加完毕,25℃继续反应3h,至出现白色浑浊,加入乙醚20ml继续沉淀,离心,40℃真空干燥,质谱检测M+Na:255.,得到化合物X-3;
Q3、向式X-3所示的化合物1.73g(6.78mmol)中加入氟氢化钾溶液(3M)4ml、盐酸(4M)4ml、水2ml、N,N-二甲基甲酰胺2ml,45℃下反应2h,将上述反应液进行TLC检测,以乙酸乙酯∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.3,用乙酸乙酯萃取,旋干,经硅胶柱层析纯化,质谱检测:177(M-F-),219(M+Na+),415(2M+Na+),得到上述式A-I-1所示的化合物;
Q4、取炔雌醇50.4mg(0.17mmol)溶解于1.5ml的N,N-二甲基甲酰胺中,将式A-I-1所示的化合物40.5mg(0.22mmol)加入上述的炔雌醇溶液中,然后加入CuSO4·5H2O8.44mg(0.034mmol)的水溶液300μL、NaAsC 66.8mg(0.34mmol)的水溶液1000μL和三(2-苯并咪唑基甲基)胺7.2mg(0.017mmol)的水溶液200μL,在45℃下反应2h,将所得反应液旋干,加入N,N-二甲基甲酰胺30ml并离心,收集上清液,即得式A-II-a所示的化合物;
Q5、将所述式A-II-a所示的化合物1.23mg(0.0025mmol)溶于N,N-二甲基甲酰胺100μL中,配制成25nM/μL溶液,然后用300μL哒嗪-盐酸-N,N-二甲基甲酰胺(pH=2.5)缓冲液将200mCi的18F-溶液经QMA柱洗脱,收集,取20μL式A-II-a所示的化合物加入到收集的18F-溶液中,在90℃下反应20min,将所得反应液用水稀释,然后负载C18柱纯化,用水洗涤三次,每次10mL,经C18柱纯化后,放化纯>98%,即得所述式(I-a)所示的化合物。
将上述制备得到的标记前体A-II-a进行1H-NMR分析,其图谱如图2所示。
实施例2
本实施例所述的具有式(I)所示结构18F标记的炔雌醇,如图1所示,,其中R1为亚乙基,具有式(I-a)结构:
化合物(I-a)的合成路线为:
上述式I-a所示的化合物的合成步骤包括:
Q1、将式X-1所示的化合物2-(二甲氨基)溴乙烷氢溴酸盐4.66g(20mmol)与叠氮化钠4.9g(76mmol)混合,加水100ml溶解,氮气保护,93℃下反应15h,反应结束加入Na2CO3将所得反应液调节pH值至中性,将上述反应液进行TLC检测,以二氯甲烷∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.5,采用二氯甲烷萃取,所得有机相经无水硫酸钠干燥,旋干,得油状液体,即为式X-2所示的化合物;
Q2、将式X-2所示的化合物1484.0mg(13mmol)加入20ml乙醚溶解,氮气保护,然后逐滴加入溴甲基硼酸频哪醇酯3158.9mg(14.3mmol)的乙醚溶液10ml,滴加完毕,20℃继续反应4h,至出现白色浑浊,加入乙醚40ml继续沉淀,离心,42℃真空干燥,得到化合物X-3;
Q3、向式X-3所示的化合物2.552g(10mmol)中加入氟氢化钾(3M)6ml、盐酸(4M)6ml、水3ml、二甲亚砜3ml,42℃下反应3h,将上述反应液进行TLC检测,以乙酸乙酯∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.3,用乙酸乙酯萃取,旋干,经硅胶柱层析纯化,得到上述式A-I-1所示的化合物;
Q4、取炔雌醇118.5mg(0.4mmol)溶解于4ml的甲醇中,将式A-I-1所示的化合物81.0mg(0.44mmol)加入上述的炔雌醇溶液中,然后加入CuSO4·5H2O22.0mg(0.088mmol)的水溶液600μL、NaAsC 142.6mg(0.72mmol)的水溶液2000μL和三(2-苯并咪唑基甲基)胺20.33mg(0.048mmol)的水溶液400μL,控制上述反应体系中甲醇与H2O体积比为1∶1.1,在40℃下反应1.5h,将所得反应液旋干,加入甲醇溶液70ml并离心,收集上清液,即得式A-II-a所示的化合物;
Q5、将所述式A-II-a所示的化合物2.26mg(0.0050mmol)溶于N,N-二甲基甲酰胺200mL中,配制成25nM/μL溶液,然后用500μL哒嗪-盐酸-N,N-二甲基甲酰胺(pH=2.5)缓冲液将200mCi的18F-溶液经QMA柱洗脱,收集,取20μL式A-II-a所示的化合物加入到收集的18F-溶液中,在90℃下反应20min,将所得反应液用水稀释,然后负载C18柱纯化,用水洗涤三次,每次10mL,即得所述式(I-a)所示的化合物。
实施例3
本实施例所述的具有式(I)所示结构18F标记的炔雌醇,如图1所示,其中R1为亚乙基,具有式(I-a)结构:
化合物(I-a)的合成路线为:
上述式I-a所示的化合物的合成步骤包括:
Q1、将式X-1所示的化合物2-(二甲氨基)溴乙烷氢溴酸盐3493.5mg(15mmol)与叠氮化钠4095.6mg(63mmol)混合,加水75ml溶解,氮气保护,87℃下反应17h,反应结束加入Na2CO3将所得反应液调节pH值至中性,将上述反应液进行TLC检测,以二氯甲烷∶甲醇=10∶1(v/v)展开,显色剂为碘单质,Rf=0.5,采用二氯甲烷萃取,所得有机相经无水硫酸钠干燥,旋干,得油状液体,即为式X-2所示的化合物;
Q2、将式X-2所示的化合物1141.5mg(10mmol)加入15ml乙醚溶解,氮气保护,然后逐滴加入溴甲基硼酸频哪醇酯3556.5mg(16.1mmol)的乙醚溶液5ml,滴加完毕,30℃继续反应2h,至出现白色浑浊,加入乙醚30ml继续沉淀,离心,41℃真空干燥,得到化合物X-3;
Q3、向式X-3所示的化合物2041.6mg(8mmol)中加入氟氢化钾(3M)5ml、盐酸(4M)5ml、水2.5ml、乙腈2.5ml,47℃下反应1h,将上述反应液进行TLC检测,以乙酸乙酯∶甲醇=10∶1(v/v)展开,显色剂碘单质,Rf=0.3,用乙酸乙酯萃取,旋干,经硅胶柱层析纯化,得到上述式A-I-1所示的化合物;
Q4、取炔雌醇88.9mg(0.3mmol)溶解于2.2ml二甲亚砜中,将式A-I-1所示的化合物49.7mg(0.27mmol)加入上述的炔雌醇溶液中,然后加入CuSO4·5H2O13.5mg(0.054mmol)的水溶液450μL、NaAsC 107.0mg(0.54mmol)的水溶液3000μL和三(2-苯并咪唑基甲基)胺9.8mg(0.024mmol)的水溶液300μL,控制上述反应体系中二甲亚砜与H2O体积比为1∶1,在50℃下反应0.5h,将所得反应液旋干,加入二甲亚砜45ml并离心,收集上清液,即得式A-II-a所示的化合物;
Q5、将所述式A-II-a所示的化合物3.69mg(0.0075mmol)溶于N,N-二甲基甲酰胺300mL中,配制成25nM/μL溶液,然后用900μL哒嗪-盐酸-N,N-二甲基甲酰胺(pH=2.5)缓冲液将600mCi的18F-溶液经QMA柱洗脱,收集,取20μL式A-II-a所示的化合物加入到收集的18F-溶液中,在90℃下反应20min,将所得反应液用水稀释,然后负载C18柱纯化,用水洗涤三次,每次10mL,即得所述式(1-a)所示的化合物。
实施例4
本实施例所述的具有式(I)所示结构18F标记的炔雌醇,如图1所示,其中R1为3,6,9,12-四氧杂十四烷亚基,具有式(I-b)结构:
化合物(I-b)的合成路线为:
上述式I-a所示的化合物的合成步骤包括:
F1、室温下,将式Y-1所示的四聚乙二醇4464.8mg(23mmol)溶解20mL四氢呋喃中,加入KOH 2917.5mg(52mmol)水溶液3mL,缓慢滴加含对甲苯磺酰氯4385.0mg(23mmol)的四氢呋喃溶液30mL,滴加完毕,置于冰浴中反应2h,反应完毕采用二氯甲烷萃取,经柱层析分离纯化(二氯甲烷/甲醇=30/1,v/v),得到油状物,即式Y-2所示的化合物;
F2、将式Y-2所示的化合物3481.2mg(10mmol)与叠氮化钠1800.0mg(30mmol)溶于20mL乙腈中,在95℃下加热反应16h,反应结束采用二氯甲烷萃取,旋蒸,得到油状物,即式Y-3所示的化合物;
F3、将式Y-3所示的化合物1468.1g(6.7mmol)溶于20mL二氯甲烷中,加入4-二甲氨基吡啶357.2mg(2.9mmol)和三乙胺8.9mL(67mmol),冰浴下搅拌,缓慢滴加含对甲苯磺酰氯3813.0g(20mmol)的二氯甲烷溶液20mL,滴加完毕,室温反应过夜,分别用1M盐酸和饱和食盐水洗涤各洗涤三次,有机相旋蒸,经柱层析分离纯化(正己烷/乙酸乙酯=2/1,v/v),得到黄色油状物,即式Y-4所示的化合物;
F4、将式Y-4所示的化合物3208.9g(8.6mmol)、N,N-二甲基乙醇胺772μL(668.1mg,7.5mmol)、KOH 1627.1mg(29mmol)溶于50mL四氢呋喃中,在70℃加热反应12h,反应结束后过滤,收集滤液,旋蒸,用二氯甲烷萃取,有机相旋干,经柱层析分离纯化(二氯甲烷/甲醇=10/1,v/v),得式Y-5所示的化合物;
F5、将式Y-5所示的化合物480mg(1.6mmol)溶于10mL干燥的四氢呋喃中,将溴甲基硼酸频那醇酯279.4μL(352.0mg,1.6mmol)溶于5mL干燥的四氢呋喃中,然后将上述溴甲基硼酸频那醇酯的四氢呋喃溶液逐滴加入上述式Y-5所示的化合物的四氢呋喃溶液中,在室温、氮气保护的条件下反应3h,将四氢呋喃旋干,得到式Y-6所示的化合物;
F6、将式Y-6所示的化合物478mg(1.1mmol)溶于1mLN,N-二甲基甲酰胺中,加入1mL氟氢化钾水溶液(4M)、1mL盐酸(3M),氮气保护,在45℃反应2h,加入10μL氨水淬灭,调节pH值至中性,旋干,经柱层析分离纯化(二氯甲烷/甲醇=20/1,v/v),得到上述式A-I-2所示的化合物;
F7、取炔雌醇53.3mg(0.18mmol)溶解于4mL N,N-二甲基甲酰胺/水(1/1,v/v)中,将式A-I-2所示的化合物66mg(0.18mmol)加入上述的炔雌醇溶液中,然后加入CuSO4·5H2O9mg(0.036mmol)、NaAsC 71.3mg(0.36mmol)和三(2-苯并咪唑基甲基)胺7.2mg(0.018mmol),控制上述反应体系中N,N-二甲基甲酰胺与H2O体积比为1∶0.9,在45℃下反应2h,将所得反应液旋干,加入N,N-二甲基甲酰胺30mL并离心,取上清液,即得式A-II-b所示的化合物;
F8、将所述式A-II-b所示的化合物1.67mg(0.0025mmol)溶于N,N-二甲基甲酰胺100μL中,配制成25nM/μL溶液,然后用300μL哒嗪-盐酸-N,N-二甲基甲酰胺(pH=2.5)缓冲液将200mCi的18F-溶液经QMA柱洗脱,收集,取20μL式A-II-b所示的化合物加入到收集的18F-溶液中,在85℃下反应30min,用水稀释,然后负载C18柱纯化,用水洗涤三次,每次10mL,即得所述式(I-a)所示的化合物。
将上述制备的标记前体A-II-b进行1H-NMR分析,其图谱分别如图3所示。
实施例5
本实施例所述的具有式(I)所示结构18F标记的炔雌醇,如图1所示,其中R1为3,6,9,12-四氧杂十四烷亚基,具有式(I-b)结构:
化合物(I-b)的合成路线为:
上述式I-a所示的化合物的合成步骤包括:
F1、室温下,将式Y-1所示的四聚乙二醇9076mg(50mmol)溶解于40mL二氧六环中,加入NaOH 4001mg(100mmol)水溶液6mL,缓慢滴加含对甲苯磺酰氯9532.5mg(50mmol)的二氧六环溶液60mL,滴加完毕,置于冰浴中反应2.5h,反应完毕采用二氯甲烷萃取,经柱层析分离纯化(二氯甲烷/甲醇=30/1,v/v),旋干,得到油状物,即式Y-2所示的化合物;
F2、将式Y-2所示的化合物5221.8mg(15mmol)与叠氮化钠2700.5mg(45mmol)溶于30mL二甲亚砜中,在97℃下加热反应17h,反应结束采用二氯甲烷萃取,旋蒸,得到油状物,即式Y-3所示的化合物;
F3、将式Y-3所示的化合物1095.6mg(5mmol)溶于15mL乙腈中,加入4-二甲氨基吡啶258.7mg(2.1mmol)和三乙胺6.6mL(50mmol),冰浴下搅拌,缓慢滴加含对甲苯磺酰氯2859.8mg(15mmol)的乙腈溶液15mL,滴加完毕,室温反应过夜,分别用1M盐酸和饱和食盐水洗涤各洗涤三次,有机相旋蒸,经柱层析分离纯化(正己烷/乙酸乙酯=2/1,v/v),得到黄色油状物,即式Y-4所示的化合物;
F4、将式Y-4所示的化合物2238.8mg(6mmol)、N,N-二甲基乙醇胺535.3μL(463.2mg,5.2mmol)、KOH1133.4mg(20.2mmol)溶于35mL四氢呋喃中,在65℃下加热反应12h,反应结束后过滤,收集滤液,旋蒸,用二氯甲烷萃取,有机相旋干,经柱层析分离纯化(二氯甲烷/甲醇=10/1,v/v),得式Y-5所示的化合物;
F5、将式Y-5所示的化合物300mg(1mmol)溶于5mL干燥的乙腈中,将溴甲基硼酸频那醇酯174.6μL(220mg,1mmol)溶于2.5mL干燥的乙腈中,然后将上述溴甲基硼酸频那醇酯的乙腈溶液逐滴加入上述式Y-5所示的化合物的乙腈溶液中,在室温、氮气保护的条件下反应3.5h,将乙腈旋干,得到式Y-6所示的化合物;
F6、将式Y-6所示的化合物217.3mg(0.5mmol)溶于0.5mL乙醇中,加入0.5mL氟氢化钾水溶液(4M)、0.5mL盐酸(3M),氮气保护,在47℃反应1.5h,加入5μL氨水淬灭,调节pH值至中性,旋干,经柱层析分离纯化(二氯甲烷/甲醇=20/1,v/v),得到上述式A-I-2所示的化合物;
F7、取炔雌醇44.4mg(0.15mmol)溶解于4mL丙酮/水(1/1,v/v)中,将式A-I-2所示的化合物71.5mg(0.195mmol)加入上述的炔雌醇溶液中,然后加入CuSO4·5H2O 6.75mg(0.027mmol)、NaAsC 53.5mg(0.27mmol)和三(2-苯并咪唑基甲基)胺4.9mg(0.12mmol),控制上述反应体系中丙酮与H2O体积比为1∶1,在50℃下反应0.5h,将所得反应液旋干,加入丙酮25mL并离心,取上清液,即得式A-II-b所示的化合物;
F8、将所述式A-II-b所示的化合物1.34mg(0.0020mmol)溶于N,N-二甲基甲酰胺80μL中,配制成25nM/μL溶液,然后用240μL哒嗪-盐酸-N,N-二甲基甲酰胺(pH=2.5)缓冲液将160mCi的18F-溶液经QMA柱洗脱,收集,取20μL式A-II-b所示的化合物加入到收集的18F-溶液中,在80℃下反应30min,用水稀释,然后负载C18柱纯化,用水洗涤三次,每次10mL,即得所述式(I-b)所示的化合物。
实施例6
本实施例所述的具有式(I)所示结构18F标记的炔雌醇,如图1所示,其中R1为3,6,9,12-四氧杂十四烷亚基,具有式(I-b)结构:
化合物(I-b)的合成路线为:
上述式I-a所示的化合物的合成步骤包括:
F1、室温下,将式Y-1所示的四聚乙二醇1941.2mg(10mmol)溶解10mLN,N-二甲基甲酰胺中,加入KOH 1268.5mg(22.6mmol)水溶液1.5mL,缓慢滴加含对甲苯磺酰氯1906.5mg(10mmol)的N,N-二甲基甲酰胺溶液15mL,滴加完毕,置于冰浴中反应1.5h,反应完毕采用二氯甲烷萃取,经柱层析分离纯化(二氯甲烷/甲醇=30/1,v/v),旋干,得到油状物,即式Y-2所示的化合物;
F2、将式Y-2所示的化合物1740.6mg(5mmol)与叠氮化钠900.0mg(15mmol)溶于10mLN,N-二甲基甲酰胺中,在93℃下加热反应15h,反应结束采用二氯甲烷萃取,旋蒸,得到油状物,即式Y-3所示的化合物;
F3、将式Y-3所示的化合物657.4mg(3mmol)溶于10mL二甲亚砜中,加入4-二甲氨基吡啶160.1g(1.3mmol)和三乙胺4.0mL(30mmol),冰浴下搅拌,缓慢滴加含对甲苯磺酰氯1715.9mg(9.0mmol)的二甲亚砜溶液10mL,滴加完毕,室温反应过夜,分别用1M盐酸和饱和食盐水洗涤各洗涤三次,有机相旋蒸,经柱层析分离纯化(正己烷/乙酸乙酯=2/1,v/v),得到黄色油状物,即式Y-4所示的化合物;
F4、将式Y-4所示的化合物1492.5mg(4mmol)、N,N-二甲基乙醇胺360.3μL(311.8mg,3.5mmol)、KOH 756.8mg(13.5mmol)溶于25mL二甲亚砜中,在67℃加热反应10h,反应结束后过滤,收集滤液,旋蒸,用二氯甲烷萃取,有机相旋干,经柱层析分离纯化(二氯甲烷/甲醇=10/1,v/v),得式Y-5所示的化合物;
F5、将式Y-5所示的化合物900mg(3mmol)溶于20mL干燥的N,N-二甲基甲酰胺中,将溴甲基硼酸频那醇酯523.9μL(660mg,3mmol)溶于10mL干燥的的N,N-二甲基甲酰胺中,然后将上述溴甲基硼酸频那醇酯的N,N-二甲基甲酰胺溶液逐滴加入上述式Y-5所示的化合物的N,N-二甲基甲酰胺溶液中,在室温、氮气保护的条件下反应3h,将N,N-二甲基甲酰胺旋干,得到式Y-6所示的化合物;
F6、将式Y-6所示的化合物869.2mg(2.0mmol)溶于2mL乙腈中,加入2mL氟氢化钾水溶液(4M)、2mL盐酸(3M),氮气保护,在42℃反应1.5h,加入20μL氨水淬灭,调节pH值至中性,旋干,经柱层析分离纯化(二氯甲烷/甲醇=20/1,v/v),得到上述式A-I-2所示的化合物;
F7、取炔雌醇59.2mg(0.2mmol)溶解于5mL乙腈/水(1/1,v/v)中,将式A-I-2所示的化合物66.0mg(0.18mmol)加入上述的炔雌醇溶液中,然后加入CuSO4·5H2O 11.0mg(0.044mmol)、NaAsC 87.2mg(0.044mmol)和三(2-苯并咪唑基甲基)胺9.8mg(0.024mmol),控制上述反应体系中乙腈与H2O体积比为1∶1.1,在40℃下反应1.0h,将所得反应液旋干,加入乙腈30mL并离心,取上清液,即得式A-II-b所示的化合物;
F8、将所述式A-II-b所示的化合物2.01mg(0.0030mmol)溶于N,N-二甲基甲酰胺120μL中,配制成25nM/μL溶液,然后用450μL哒嗪-盐酸-N,N-二甲基甲酰胺(pH=2.5)缓冲液将300mCi的18F-溶液经QMA柱洗脱,收集,取20μL式A-II-b所示的化合物加入到收集的18F-溶液中,在85℃下反应30min,用水稀释,然后负载C18柱纯化,用水洗涤三次,每次15mL,即得所述式(I-b)所示的化合物。
实验例
一、标记前体和产物的纯度分析
1.标记前体的纯度分析
在280nm紫外波长下,将实施例1制备的标记前体A-II-a、实施例4制备的标记前体A-II-b分别进行液相分析,结果如图4、图5所示,采用实施例1标记前体A-II-a、实施例4制备标记前体A-II-b的高效液相检测显示均为一单峰。其中,标记前体A-II-a纯度为99%,保留时间TR为16.1min;标记前体A-II-b纯度为99%,保留时间TR为16.5min。
2.标记产物的纯度分析
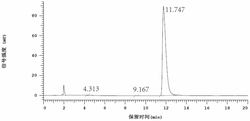
在280nm紫外波长下,将实施例1制备的标记产物I-a、实施例4制备的标记产物I-b分别进行放射性高效液相分析,结果如图6、图7所示,其中图6为标记产物I-a纯化后Radio-HPLC分析图谱,图7为标记产物I-b纯化后Radio-HPLC分析图谱,采用实施例1制备的标记产物I-a、实施例4制备的标记产物I-b的放射性高效液相检测显示均为一单峰。其中,标记产物I-a纯度为99%,保留时间TR为16.3min;标记产物I-b纯度为98%,保留时间TR为16.6min。
将实施例1制备的标记前体A-II-a、实施例4制备的标记前体A-II-b分别溶于DMF溶液,配制25nM/μL的溶液。采用200mCi的18F-用300μL的Pry-HCl-DMF(pH=2.5)的缓冲液从QMA柱上洗脱下来,分别用EP管收集,将标记前体A-II-a、A-II-b的DMF溶液分别加入到上述含18F-的EP管中,不同温度下分别加热20min。反应混合物用水稀释后负载到C18柱上,用水(3×10m1)洗涤。纯的放射性标记产物用乙醇与水(1∶1)洗脱到小玻璃瓶中。取一点样品通过HPLC进行质量控制分析。所得标记率如图8、图9所示。80℃是一个有效标记温度并用于后续实验。经C18柱纯化后,放化纯>98%。
二、体外稳定性测试
1.标记前体的体外稳定性测试
将实施例1制备的标记前体A-II-a、实施例4制备的标记前体A-II-b分别与哒嗪-盐酸(pH 2.0-2.5)的缓冲液一起在不同温度下加热20min,用HPLC检测其纯度,结果如图10、图11所示。
2.标记产物的体外稳定性测试
取100μCi放射性的实施例1制备的标记产物I-a、实施例4制备的标记产物I-b分别加入到500μL PBS或血清(该血清Biological Industries提供)中,并37℃分别孵育0,1,2,4h。在预定时间点,取出PBS混合液中一部分,直接进行HPLC分析;对于血清样品,取70%MeCN加至血清中,10000g离心5min,取上清用HPLC分析。结果显示,在4h后,标记产物I-a的放化纯仍大于97%。其中,图12为标记产物I-a在PBS中孵育4h后radio-HPLC分析图谱,图13为标记产物I-a在血清中孵育4h后radio-HPLC分析图谱,图14为标记产物I-a在PBS和血清中孵育不同时间的放化纯图谱;图15为标记产物I-b在PBS中孵育4h后radio-HPLC分析图谱,图16为标记产物I-b在血清中孵育4h后radio-HPLC分析图谱,图17为标记产物I-b在PBS和血清中孵育不同时间的放化纯图谱。
三、脂水分配系数
1.标记产物I-a脂水分配系数测定
将PBS缓冲液与等体积的正辛醇充分振荡,使两相相互饱和,室温下静置24h以上,用分液漏斗将两相分离。取实施例1制备的标记产物I-a溶液0.05ml,加入1mL正辛醇、0.95mL PBS缓冲液混合,用涡旋混合器振荡5min,充分混匀,然后4000g离心5min,在有机相和水相中分别取100μL样品于γ计数管中,用γ计数仪测定其放射性活度,实验重复两次。计算脂水分配系数的log P值。用公式log P=log(CPM水相/CPM有机相)计算,实际得到:log P=0.52±0.09,为亲脂性。
2.标记产物I-b脂水分配系数测定
将PBS缓冲液与等体积的正辛醇充分振荡,使两相相互饱和,室温下静置24h以上,用分液漏斗将两相分离。取实施例2制备的标记产物I-b溶液0.05ml,加入1mL正辛醇、0.95mL PBS缓冲液混合,用涡旋混合器振荡5min,充分混匀,然后4000g离心5min,在有机相和水相中分别取100μL样品于γ计数管中,用γ计数仪测定其放射性活度,实验重复两次。计算脂水分配系数的log P值。用公式log P=log(CPM水相/CPM有机相)计算,实际得到:log P=-0.176±0.028,为亲水性。
四、细胞毒性实验
以6000个细胞/孔的密度将T47D细胞(所述细胞由南京科佰生物科技有限公司提供)接种到96孔板中,培养基体积100μL/孔,37℃过夜培养12h,吸除原有培养基后每孔加入200μL的含药浓度为12.5、25、50、100μM培养基,每个浓度设置至少6个复孔,然后分别放入培养箱分别培养3h,6h,12h,24h,达到培养时间时,每孔加入20μL的MTT(5mg/mL PBS)溶液,放入培养箱中继续培养4h后,小心吸除上清液后,每孔加入150μL的DMSO,放置在摇床上震荡至孔底紫色结晶完全溶解,用酶标仪测定490nm波长下的吸光度OD值,然后计算细胞生存率。标记前体A-II-a、A-II-b细胞毒性实验结果分别如图18、19所示,在100μM药物浓度下,T47D细胞生存率分别为89.2%、88.9%,说明标记前体A-II-a、A-II-b对细胞是无毒的。
五、细胞摄取与阻断
1.标记产物I-a
将T47D乳腺癌细胞接种在24孔板,密度为2×105细胞/孔,过夜培养。T47D细胞由实施例1制备的标记产物I-a在37℃下进行培养15,30,60,120min,每孔0.5μCi。在阻断特异性结合的实验中,将雌二醇加入到24孔板的T47D细胞中,终浓度5μM。培养结束后,T47D细胞用PBS洗三遍,用0.1M的NaOH裂解,收集裂解液,保留的放射性采用γ计数器测量。细胞摄取采用加入的剂量百分率表述(%AD),该剂量经衰变校正。所有的实验均三个复孔重复两次。
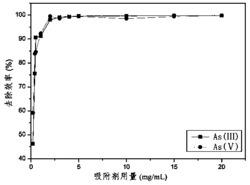
实验结果如图20所示,细胞总摄取率为3.5%,并能够被雌二醇阻断至1.7%,证明此标记产物I-a对T47D细胞具有靶向性。
2.标记产物I-b
将T47D乳腺癌细胞接种在24孔板,密度为2×105细胞/孔,过夜培养。T47D细胞由实施例4制备的标记产物I-b在37℃下进行培养15,30,60,120min,每孔0.5μCi。在阻断特异性结合的实验中,将雌二醇加入到24孔板的T47D细胞中,终浓度5μM。培养结束后,T47D细胞用PBS洗三遍,用0.1M的NaOH裂解,收集裂解液,保留的放射性采用γ计数器测量。细胞摄取采用加入的剂量百分率表述(%AD),该剂量经衰变校正。所有的实验均三个复孔重复两次。
实验结果如图21所示,细胞总摄取率为1.61%,并能够被雌二醇阻断至0.94%,证明此标记产物I-b对T47D细胞具有靶向性。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
一种18F标记的炔雌醇及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0