

IPC分类号 : C07J63/00I,A61K31/56I,A61P31/04I




专利摘要
本发明提供了一种C3和C20双酯化的甘草次酸衍生物,具有式Ⅰ所示结构,或其药学上可接受的盐、溶剂化物、光学异构体或多晶型物。实验结果表明,本发明提供的C3和C20双酯化的甘草次酸衍生物对金黄色葡萄球菌具有很好的抑菌作用,能够抑制金黄色葡萄球菌ATCC6538、金黄色葡萄球菌ATCC12228和金黄色葡萄球菌ATCC29213,为金黄色葡萄球菌的抗感染药物提供了一种新的选择。
权利要求
1.一种C3和C20双酯化的甘草次酸衍生物,其特征在于,具有式Ⅰ所示结构,或其药学上可接受的盐或光学异构体:
其中,R为乙基;
R'为对甲基苯基或邻甲氧基苯基。
2.权利要求1所述的C3和C20双酯化的甘草次酸衍生物的制备方法,其特征在于,包括以下步骤:
a)将式Ⅱ所示的甘草次酸和苯酸酐类化合物或苯酰氯类化合物混合,在4-二甲氨基吡啶的催化作用下,进行第一酯化反应,得到式Ⅲ所示的C3被酯化的衍生物;
b)将式Ⅲ所示的衍生物在强酸性条件下,加入醇类化合物,进行第二酯化反应,得到C3和C20被酯化的式Ⅰ所示化合物;
或者包括以下步骤:
c)将式Ⅱ所示的甘草次酸在强酸性条件下,加入醇类化合物,进行第三酯化反应,得到式Ⅳ所示的C20被酯化的衍生物;
d)将式Ⅳ所示的衍生物和苯酸酐类化合物或苯酰氯类化合物混合,在4-二甲氨基吡啶的催化作用下,进行第四酯化反应,得到C3和C20被酯化的式Ⅰ所示化合物;
所述苯酸酐类化合物选自对甲基苯基酸酐或邻甲氧基苯基酸酐;
所述苯酰氯类化合物选自对甲基苯基酰氯或邻甲氧基苯甲酰氯;
所述醇类化合物为乙醇;
其中,R为乙基;
R'为对甲基苯基或邻甲氧基苯基。
3.根据权利要求2所述的制备方法,其特征在于,所述步骤a)所述的第一酯化反应的温度为10℃~40℃;所述步骤b)所述的第二酯化反应的温度为10℃~40℃;所述步骤c)所述的第三酯化反应的温度为10℃~40℃;所述步骤d)所述的第四酯化反应的温度为10℃~40℃。
4.根据权利要求2所述的制备方法,其特征在于,所述步骤a)中,所述甘草次酸和苯酸酐类化合物的摩尔比为1:1.5~3.0;所述甘草次酸和苯酰氯类化合物的摩尔比为1:2.0~4.0;所述步骤b)中,所述式Ⅲ所示的衍生物与醇类化合物的摩尔比为1:2.0~4.5;
所述步骤c)中,所述甘草次酸和醇类化合物的摩尔比为1:2.0~4.5;所述步骤d)中,所述式Ⅳ所示的衍生物和苯酸酐类化合物的摩尔比为1:1.5~3.0;所述式Ⅳ所示的衍生物和苯酰氯类化合物的摩尔比为1:2.0~4.0。
5.权利要求1所述的C3和C20双酯化的甘草次酸衍生物或权利要求2~4任一项所述的制备方法制备的C3和C20双酯化的甘草次酸衍生物在制备抗葡萄球菌制剂中的应用。
6.一种抗葡萄球菌制剂,包括权利要求1所述的C3和C20双酯化的甘草次酸衍生物或权利要求2~4任一项所述的制备方法制备的C3和C20双酯化的甘草次酸衍生物。
说明书
技术领域
本发明涉及药物合成技术领域,尤其涉及一种C3和C20双酯化的甘草次酸衍生物及其制备方法和应用。
背景技术
金黄色葡萄球菌是全世界发病和死亡的重要原因。这种普遍存在的革兰氏阳性细菌可引起轻度至危及生命的疾病,常见的临场症状为,化脓性疾病,食物中毒,肺炎和败血症。化学疗法通常用于金黄色葡萄球菌感染性疾病。但是,耐甲氧西林的金黄色葡萄球菌(MRSA)的出现代表了治疗金黄色葡萄球菌感染的问题变得十分严峻。耐多药病原体的感染引起的发病率和死亡率的增加,突出了对开发新型抗菌剂以保护人类和植物生命健康的迫切需求。
目前被设计为新型抗菌剂的已批准药物中,有很大一部分是天然产物或衍生自天然产物支架。天然产物及其具有独特而多样的化学结构的衍生物通常具有广谱杀菌活性,特别是对抗生素耐药菌株的抑制活性十分显著,这为新型抗菌疗法提供了合理设计开发思路。
甘草次酸是甘草提取物中的主要成分。先前的研究表明,甘草次酸具有许多特性,包括抗炎,抗过敏,抗消化性溃疡和抗病毒活性。甘草次酸对某些细菌具有抗菌作用,但该活性的潜在机制尚不清楚。为了进一步探究甘草次酸的抗菌机制,有必要对甘草次酸进行改造,提供更多有用的活性数据。
发明内容
有鉴于此,本发明要解决的技术问题在于提供一种C3和C20双酯化的甘草次酸衍生物及其制备方法和应用,用于进一步探究甘草次酸对革兰氏阳性菌细菌的作用。
为解决以上技术问题,本发明提供了一种C3和C20双酯化的甘草次酸衍生物,具有式Ⅰ所示结构,或其药学上可接受的盐、溶剂化物、光学异构体或多晶型物:
其中,R为C1~C10烷基;
R'为C1~C10烷基、取代或非取代的五元或六元芳香基团。
本发明对甘草次酸的3号碳原子(C3)和20号碳原子(C20)进行酯化修饰。
其中,C3修饰的酯化基团中的R'优选为C1~C10烷基、取代或非取代的五元或六元芳香基团。
所述C1~C10烷基进一步优选为C1~C5烷基,更优选为甲基、乙基、丙基或正丁基。
本发明中,所述五元芳香基团指包含碳原子以及杂原子在内的五元芳香环或芳香杂环基团;所述六元芳香基团指包含碳原子以及杂原子在内的六元芳香环或芳香杂环基团。
所述取代或非取代的五元或六元芳香基团中,取代基优选为C1~C5烷基或C1~C5烷氧基;所述五元或六元芳香基团优选为苯基、呋喃基、吡咯基或噻吩基。
进一步优选的,所述取代或非取代的五元或六元芳香基团为苯基、对甲基苯基、邻甲基苯基、间甲基苯基、对甲氧基苯基、邻甲氧基苯基或间甲氧基苯基。
C20修饰的酯化基团中的R优选为C1~C10烷基,进一步优选为C1~C5烷基,更优选为甲基、乙基、丙基或正丁基。
本发明公开了上述C3和C20双酯化的甘草次酸衍生物的制备方法,包括以下步骤:
a)将式Ⅱ所示的甘草次酸和酸酐类化合物、苯酸酐类化合物或苯酰氯类化合物混合,在4-二甲氨基吡啶的催化作用下,进行第一酯化反应,得到式Ⅲ所示的C3被酯化的衍生物;
b)将式Ⅲ所示的衍生物在强酸性条件下,加入醇类化合物,进行第二酯化反应,得到C3和C20被酯化的式Ⅰ所示化合物;
或者包括以下步骤:
c)将式Ⅱ所示的甘草次酸在强酸性条件下,加入醇类化合物,进行第三酯化反应,得到式Ⅳ所示的C20被酯化的衍生物;
d)将式Ⅳ所示的衍生物和酸酐类化合物、苯酸酐类化合物或苯酰氯类化合物混合,在4-二甲氨基吡啶的催化作用下,进行第四酯化反应,得到C3和C20被酯化的式Ⅰ所示化合物;
其中,R为C1~C10烷基;
R'为C1~C10烷基、取代或非取代的五元或六元芳香基团。
本发明中,步骤a)、b)的反应方程式如下:
首先,以式Ⅱ所示的甘草次酸为原料,将甘草次酸溶于有机溶剂中,常温下加入酸酐类化合物、苯酸酐类化合物或苯酰氯类化合物,在4-二甲氨基吡啶的催化作用下,进行第一酯化反应,得到式Ⅲ所示的C3被酯化的衍生物。
所述第一酯化反应的温度优选为10℃~40℃,反应时间优选为4~6h。
所述甘草次酸和酸酐类化合物或苯酸酐类化合物的摩尔比优选为1:1.5~3.0;所述甘草次酸和苯酰氯类化合物的摩尔比优选为1:2.0~4.0。
所述有机溶剂优选为吡啶。
然后,将式Ⅲ所示的衍生物溶于有机溶剂,在强酸性条件下,加入醇类化合物,进行第二酯化反应,得到C3和C20被酯化的式Ⅰ所示化合物。
所述第二酯化反应的温度优选为10℃~40℃,反应时间优选为6~12h。
所述式Ⅲ所示的衍生物与醇类化合物的摩尔比优选为1:2.0~4.5。
所述有机溶剂优选为甲醇或者乙醇。
本发明中,所述步骤c)、d)的反应方程式如下:
首先,以式Ⅱ所示的甘草次酸为原料,将甘草次酸溶于有机溶剂中,在强酸性条件下,加入醇类化合物,进行第三酯化反应,得到式Ⅳ所示的C20被酯化的衍生物。
所述第三酯化反应的温度优选为10℃~40℃,反应时间优选为12~24h。
所述甘草次酸和醇类化合物的摩尔比优选为1:2.0~4.5。
所述有机溶剂优选为甲醇或者乙醇。
然后,将式Ⅳ所示的衍生物溶于有机溶剂中,常温下加入酸酐类化合物、苯酸酐类化合物或苯酰氯类化合物,在4-二甲氨基吡啶的催化作用下,进行第四酯化反应,得到C3和C20被酯化的式Ⅰ所示化合物。
所述第四酯化反应的温度优选为10℃~40℃,反应时间优选为12~18h。
所述式Ⅳ所示的衍生物和酸酐类化合物或苯酸酐类化合物的摩尔比优选为1:1.5~3.0;所述式Ⅳ所示的衍生物和苯酰氯类化合物的摩尔比优选为1:2.0~4.0。
所述有机溶剂优选为吡啶。
本发明中,上述酸酐类化合物优选为甲酸酐、乙酸酐、丙酸酐或丁酸酐;所述苯酸酐类化合物优选为苯基酸酐或苯甲氧基酸酐;所述苯酰氯类化合物优选为邻甲基苯甲酰氯、间甲基苯甲酰氯、邻甲氧基苯甲酰氯或间甲氧基苯甲酰氯;所述醇类化合物优选为甲醇、乙醇、丙醇或正丁醇。
本发明中,在第二酯化反应或第四酯化反应结束后,优选的,还包括纯化步骤。
优选的,所述纯化步骤包括蒸除溶剂,通过柱层析法进行提纯。
在本发明的一些具体实施例中,所述纯化步骤具体包括:
过滤,减压蒸除溶剂,用蒸馏水萃取,干燥,收集固体层,对固体层加碱调节pH值至7~8后,水洗至中性,过滤收集固体,烘干固体后通过硅胶柱层析纯化。
本发明制备的所有衍生物均为白色固体。
本发明上述结构式中,单键表示甲基。
表示连接的位置。
本发明公开了上述C3和C20双酯化的甘草次酸衍生物或上述制备方法制备的C3和C20双酯化的甘草次酸衍生物在制备抗葡萄球菌制剂中的应用。
本发明公开了一种抗葡萄球菌制剂,包括上述C3和C20双酯化的甘草次酸衍生物或上述制备方法制备的C3和C20双酯化的甘草次酸衍生物。
本发明所述的抗葡萄球菌制剂的给药方法可以是口服给药,注射给药,喷雾吸入法,局部给药,经直肠给药,经鼻给药,含服给药,或通过植入性药盒给药等。
本发明所述的抗葡萄球菌制剂的剂型可以是口服剂型,如胶囊,片剂,丸剂,粉剂,粒剂,水制悬浮液或溶液等。
上述抗葡萄球菌制剂还可以包括药学上可接受的载体,赋形剂,稀释剂,辅剂,媒介物或它们的组合。
上述药学上可接受的载体,赋形剂,稀释剂,辅剂,媒介物可以根据本领域技术人员技术常识进行选择,本发明对此并无特殊限定。
与现有技术相比,本发明提供了一种C3和C20双酯化的甘草次酸衍生物,具有式Ⅰ所示结构,或其药学上可接受的盐、溶剂化物、光学异构体或多晶型物。实验结果表明,本发明提供的C3和C20双酯化的甘草次酸衍生物对金黄色葡萄球菌具有很好的抑菌作用,能够抑制金黄色葡萄球菌ATCC 6538、金黄色葡萄球菌ATCC 12228和金黄色葡萄球菌ATCC29213,为金黄色葡萄球菌的抗感染药物提供了一种新的选择。
附图说明
图1为本发明制备的化合物4a的高分辨质谱图;
图2为本发明制备的化合物4b的高分辨质谱图。
具体实施方式
为了进一步说明本发明,下面结合实施例对本发明提供的C3和C20双酯化的甘草次酸衍生物及其制备方法和应用进行详细描述。
实施例1化合物2a的制备
称取甘草次酸(化合物1)(470mg,1mmol)于100mL有磁子的圆底烧瓶中,然后量取20mL无水甲醇于烧瓶中,在室温下,搅拌3min待完全溶解后,用玻璃吸管吸取1.5mL的浓硫酸,缓慢的加入体系中,然后置于常温条件下进行酯化反应24h。最后在45℃下减压蒸馏除去甲醇溶剂得到化合物2a粗产物。通过硅胶柱层析法(洗脱剂:石油醚:乙酸乙酯(体积比)=2:1)得到化合物2a,为白色固体,产率为90%。
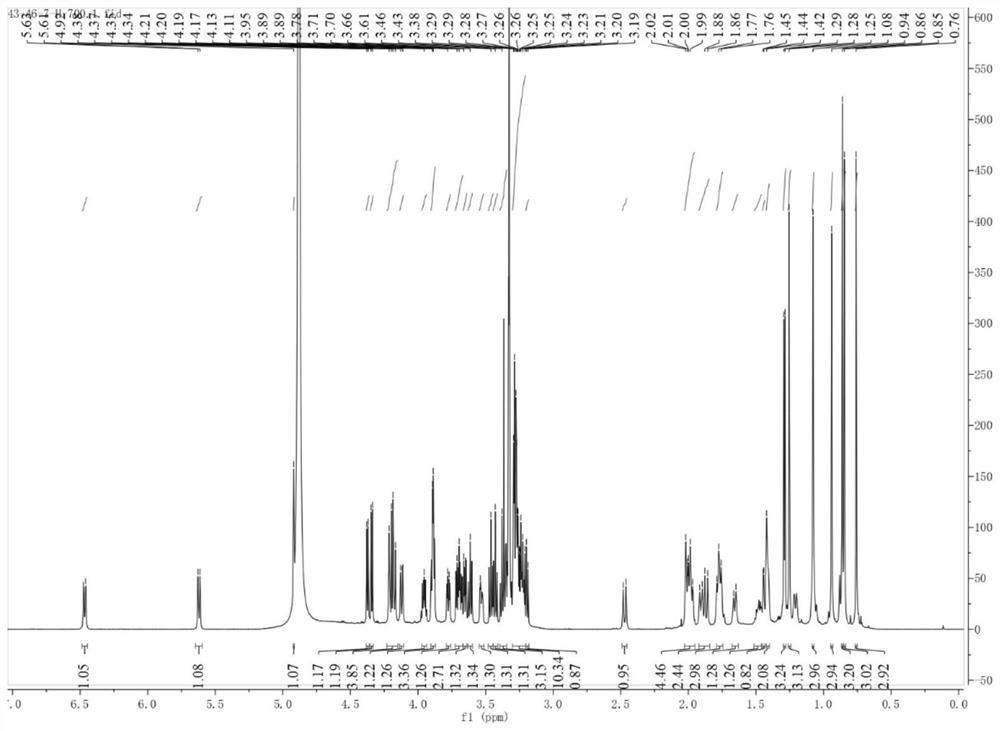
进行1HNMR 13C NMR和高分辨质谱分析,结果为:1H NMR(400MHz,CDCl3)δ5.66(s,1H),3.68(s,3H),3.27–3.17(m,1H),2.85–2.75(m,1H),2.33(s,1H),2.10–1.96(m,3H),1.94–1.87(m,1H),1.87–1.77(m,1H),1.67–1.55(m,5H),1.49(d,J=6.6Hz,2H),1.46–1.37(m,3H),1.36(s,3H),1.30(d,J=9.7Hz,2H),1.21–1.16(m,1H),1.14(s,3H),1.13(s,3H),1.12(s,3H),1.00(s,3H),0.98–0.93(m,1H),0.80(s,6H),0.72–0.67(m,1H).13C NMR(400MHz,CDCl3)δ200.22,176.94,169.17,128.57,78.78,61.84,54.98,51.79,48.41,45.40,44.06,43.21,41.13,39.15,37.76,37.11,32.79,31.85,31.16,28.53,28.34,28.11,27.33,26.50,26.44,23.41,18.69,17.50,16.38,15.59.HRMS(ESI)C31H49O4(485.3625);485.3625(M+H)+。
实施例2化合物2b的制备
称取甘草次酸(化合物1)(470.0mg,1mmol)于100mL有磁子的圆底烧瓶中,然后量取20mL无水乙醇于烧瓶中,在室温下,搅拌3min待完全溶解后,用玻璃吸管吸取1.5mL的浓硫酸,缓慢的加入体系中,然后置于常温条件下进行酯化反应24h。最后在45℃下减压蒸馏除去乙醇溶剂得到化合物2b粗产物。通过硅胶柱层析法(洗脱剂:石油醚:乙酸乙酯(体积比)=2:1)得到化合物2b为白色固体,产率为88%。
进行1H NMR 13C NMR和高分辨质谱分析,结果为:1H NMR(400MHz,CDCl3)δ5.62(s,1H),4.23–4.07(m,2H),3.25–3.17(m,1H),2.81–2.72(m,1H),2.32(s,1H),2.12–2.04(m,1H),2.04–1.93(m,2H),1.93–1.86(m,1H),1.85–1.75(m,1H),1.70–1.49(m,6H),1.46–1.38(m,2H),1.38–1.28(m,6H),1.24(t,J=7.1Hz,3H),1.17(d,J=12.6Hz,1H),1.12(s,3H),1.12(s,3H),1.10(s,3H),1.03–0.99(m,1H),0.98(s,3H),0.96–0.90(m,1H),0.78(s,6H),0.68(d,J=11.5Hz,1H).13C NMR(400MHz,CDCl3)δ200.23,176.39,169.32,128.51,78.74,61.83,60.32,54.95,48.39,45.39,43.84,43.22,41.09,39.14,37.73,37.10,32.78,31.82,31.13,28.57,28.33,28.11,27.31,26.50,26.43,23.40,18.68,17.50,16.37,15.60,14.34.HRMS(ESI)C32H51O4(499.3782);499.3787(M+H)+。
实施例3化合物3a的制备
称取甘草次酸(化合物1)(470.0mg,1mmol)于100mL有磁子的圆底烧瓶中,然后量取20mL的吡啶作为溶剂,室温下搅拌2min待完全溶解之后,加入4-二甲氨基吡啶(155.7mg,1.2mmol),待搅拌均匀后,最后加入乙酸酐(86.8mg,0.85mmol),反应6h之后,用TLC检测反应终点。在55℃下减压蒸馏除去吡啶溶剂得到化合物3a粗产物。通过硅胶柱层析法(洗脱剂:石油醚:乙酸乙酯(体积比)=2:1)得到化合物3a,为白色固体,产率为80%。
进行1H NMR 13C NMR和高分辨质谱分析,结果为:1H NMR(400MHz,CDCl3)δ5.71(s,1H),4.60–4.49(m,1H),2.86–2.76(m,1H),2.36(d,J=7.7Hz,1H),2.22–2.16(m,1H),2.05(s,3H),2.03–1.89(m,3H),1.88–1.80(m,1H),1.71–1.57(m,5H),1.47(t,J=9.1Hz,1H),1.44–1.39(m,3H),1.37(s,3H),1.26(s,3H),1.23(s,3H),1.17(s,3H),1.13(s,3H),1.05(s,1H),1.01(s,1H),0.88(s,6H),0.83(d,J=5.6Hz,3H),0.80(d,J=5.9Hz,1H).13C NMR(400MHz,CDCl3)δ200.26,180.84,171.02,169.33,128.50,80.66,61.73,55.06,48.27,45.48,43.77,43.23,40.92,38.81,38.08,37.73,36.98,32.75,31.88,30.96,29.69,28.53,28.42,28.06,26.44,23.59,23.36,21.29,18.71,17.40,16.69,16.40.HRMS(ESI)C32H48O5Na(535.3394);535.3403(M+Na)+。
实施例4化合物3b的制备
称取甘草次酸(化合物1)(470.0mg,1mmol)于100mL有磁子的圆底烧瓶中,然后量取20mL的吡啶作为溶剂,室温下搅拌2min待完全溶解之后,加入4-二甲氨基吡啶(155.7mg,1.2mmol),待搅拌均匀后,最后加入苯甲酸酐(192.3mg,0.85mmol),反应6h之后,用TLC检测反应终点。在55℃下减压蒸馏除去吡啶溶剂得到化合物3b粗产物。通过硅胶柱层析法(洗脱剂:石油醚:乙酸乙酯(体积比)=2:1)得到化合物3b,为白色固体,产率为75%。
进行1H NMR 13C NMR和高分辨质谱分析,结果为:1H NMR(400MHz,CDCl3)δ8.07–8.01(m,2H),7.55(t,J=7.4Hz,1H),7.44(t,J=7.6Hz,2H),5.73(s,1H),4.83–4.73(m,1H),2.91–2.81(m,1H),2.42(s,1H),2.24–2.17(m,1H),2.09–1.95(m,3H),1.95–1.87(m,1H),1.87–1.72(m,3H),1.72–1.59(m,3H),1.56–1.48(m,1H),1.46–1.38(m,6H),1.37–1.31(m,1H),1.26(d,J=1.4Hz,1H),1.24(s,3H),1.22(s,3H),1.18(d,J=4.7Hz,1H),1.16(s,3H),1.14–1.08(m,1H),1.04(s,3H),0.96(s,3H),0.89(d,J=10.3Hz,1H),0.85(s,3H).13CNMR(400MHz,CDCl3)δ200.27,181.11,180.97,169.37,166.29,132.71,130.98,129.55,128.52,128.31,81.29,61.74,55.15,48.28,45.51,43.79,43.27,40.93,38.83,38.48,37.74,37.04,32.76,31.89,30.97,28.55,28.43,28.24,26.46,23.66,23.40,18.74,17.44,16.98,16.43.HRMS(ESI)C37H50O5Na(597.3550);597.3565(M+Na)+。
实施例5化合物3c的制备
称取甘草次酸(化合物1)(470.0mg,1mmol)于100mL有磁子的圆底烧瓶中,然后量取20mL的吡啶作为溶剂,室温下搅拌2min待完全溶解之后,加入4-二甲氨基吡啶(155.7mg,1.2mmol),吡啶(100.7mg,1.2mmol)待搅拌均匀后,最后加入对甲基苯基酰氯(295.4mg,1.9mmol),反应6h之后,用TLC检测反应终点。在55℃下减压蒸馏除去吡啶溶剂得到化合物3c粗产物。通过硅胶柱层析法(洗脱剂:石油醚:乙酸乙酯(体积比)=3:1)得到化合物3c,为白色固体,产率为92%。
进行1H NMR 13C NMR和高分辨质谱分析,结果为:1H NMR(400MHz,CDCl3)δ7.91(s,1H),7.89(s,1H),7.30(s,1H),7.28(s,1H),5.69(s,1H),3.29–3.16(m,1H),2.83–2.71(m,1H),2.43(s,3H),2.35–2.26(m,2H),2.12–1.99(m,3H),1.91–1.81(m,1H),1.75(t,J=13.7Hz,1H),1.70–1.51(m,5H),1.51–1.39(m,4H),1.38(s,3H),1.34(s,3H),1.30–1.22(m,1H),1.17(d,J=12.1Hz,1H),1.13(s,6H),1.05(d,J=13.8Hz,1H),1.00(s,3H),0.98–0.90(m,1H),0.86(s,3H),0.80(s,3H),0.70(d,J=11.5Hz,1H).13C NMR(400MHz,CDCl3)δ200.06,172.11,168.54,162.42,145.67,130.46,129.63,128.81,126.15,78.78,61.86,54.96,48.16,45.51,45.40,43.23,40.98,39.17,39.15,37.46,37.12,32.78,32.04,30.96,28.39,28.12,27.54,27.29,26.47,26.41,23.47,21.84,18.71,17.51,16.37,15.59.HRMS(ESI)C38H52O5Na(611.3707);611.3711(M+Na)+。
实施例6化合物3d的制备
称取甘草次酸(化合物1)(470.0mg,1mmol)于100mL有磁子的圆底烧瓶中,然后量取20mL的吡啶作为溶剂,室温下搅拌2min待完全溶解之后,加入4-二甲氨基吡啶(155.7mg,1.2mmol),吡啶(100.7mg,1.2mmol)待搅拌均匀后,最后加入邻甲氧基苯基酰氯(311.5mg,1.9mmol),反应6h之后,用TLC检测反应终点。在55℃下减压蒸馏除去吡啶溶剂得到化合物3d粗产物。通过硅胶柱层析法(洗脱剂:石油醚:乙酸乙酯(体积比)=3:1)得到化合物3d,为白色固体,产率为95%。
进行1H NMR 13C NMR和高分辨质谱分析,结果为:1H NMR(400MHz,CDCl3)δ7.83(dd,J=7.8,1.7Hz,1H),7.59–7.52(m,1H),7.04(d,J=7.6Hz,1H),7.00(d,J=8.4Hz,1H),5.66(s,1H),3.90(s,3H),3.28–3.17(m,1H),2.84–2.73(m,1H),2.37–2.28(m,2H),2.12–1.95(m,3H),1.90–1.80(m,1H),1.72(t,J=11.3Hz,1H),1.66–1.54(m,5H),1.49–1.39(m,4H),1.37(s,3H),1.32(s,3H),1.24(d,J=8.6Hz,1H),1.20(d,J=12.2Hz,1H),1.13(s,6H),1.08–1.02(m,1H),1.00(s,3H),0.97–0.91(m,1H),0.86(s,3H),0.80(s,3H),0.69(d,J=11.6Hz,1H).13C NMR(400MHz,CDCl3)δ200.04,172.30,168.65,161.72,160.21,135.41,132.73,128.76,120.36,118.30,112.23,78.78,61.86,55.94,54.98,47.98,45.38,45.20,43.20,40.92,39.17,39.15,37.43,37.13,32.78,31.98,30.92,28.41,28.11,27.45,27.32,26.48,26.44,23.48,18.72,17.51,16.36,15.57.HRMS(ESI)C38H52O6Na(627.3656);627.3658(M+Na)+。
实施例7化合物4a的制备
称取化合物3c(616.3mg,1mmol)于100mL有磁子的圆底烧瓶中,然后量取20mL无水乙醇于烧瓶中,在室温下,搅拌3min待完全溶解后,用玻璃吸管吸取1.5mL的浓硫酸,缓慢的加入体系中,然后置于常温条件下进行酯化反应24h。最后在45℃下减压蒸馏除去乙醇溶剂得到化合物4a粗产物。通过硅胶柱层析法(洗脱剂:石油醚:乙酸乙酯(体积比)=15:1)得到化合物4a为白色固体,产率为77%。
进行1H NMR 13C NMR和高分辨质谱分析,结果为:1H NMR(400MHz,CDCl3)δ7.88-7.75(m,1H),7.65(s,1H),7.25(s,1H),7.20-7.15(d,1H),5.68(s,1H),4.20–4.05(m,2H)3.18–3.06(m,1H),2.80–2.75(d,1H),2.40(s,3H),2.38–2.29(m,2H),2.22–2.02(m,3H),1.95–1.86(m,1H),1.78(t,J=13.7Hz,1H),1.70–1.58(m,4H),1.55–1.42(m,5H),1.37(s,3H),1.31(s,3H),1.28–1.20(m,4H),1.15(d,J=12.1Hz,1H),1.12(s,6H),1.02(d,J=13.8Hz,1H),0.99(s,3H),0.97–0.92(m,1H),0.85(s,3H),0.82(s,3H),0.73(d,J=11.5Hz,1H).13C NMR(400MHz,CDCl3)δ201.16,170.01,165.34,160.48,146.66,132.11,124.63,127.71,125.18,78.88,62.16,60.40,55.06,47.86,45.99,45.30,42.21,40.58,39.44,39.05,37.88,37.18,32.01,31.84,30.99,28.80,28.22,27.34,27.09,26.93,26.54,23.66,21.32,18.55,17.56,16.99,16.05,14.93.HRMS(ESI)C40H51O5(616.4028);615.4062(M-H)-
图1为化合物4a的高分辨质谱图。
实施例8化合物4b的制备
称取化合物3d(632.5mg,1mmol)于100mL有磁子的圆底烧瓶中,然后量取20mL无水乙醇于烧瓶中,在室温下,搅拌3min待完全溶解后,用玻璃吸管吸取1.5mL的浓硫酸,缓慢的加入体系中,然后置于常温条件下进行酯化反应24h。最后在45℃下减压蒸馏除去乙醇溶剂得到化合物4b粗产物。通过硅胶柱层析法(洗脱剂:石油醚:乙酸乙酯(体积比)=10:1)得到化合物4b为白色固体,产率为68%。
进行1H NMR 13C NMR和高分辨质谱分析,结果为:1H NMR(400MHz,CDCl3)δ7.82–7.77(m,1H),7.48–7.42(m,1H),6.98(d,J=5.0Hz,1H),6.96(d,J=6.7Hz,1H),5.65(s,1H),4.82–4.69(m,1H),4.23–4.07(m,2H),3.89(s,3H),2.89–2.79(m,1H),2.40(s,1H),2.14–1.85(m,5H),1.85–1.75(m,3H),1.73–1.56(m,4H),1.53–1.41(m,2H),1.38(s,3H),1.36–1.30(m,2H),1.26(t,J=7.1Hz,4H),1.19(s,3H),1.14(s,3H),1.14(s,3H),1.06(d,J=18.8Hz,1H),0.99(s,3H),0.99(s,3H),0.87(d,J=10.8Hz,1H),0.80(s,3H).13C NMR(400MHz,CDCl3)δ200.16,176.41,169.36,166.09,159.22,133.26,131.58,128.50,120.85,120.07,112.00,81.15,61.76,60.33,55.82,55.13,48.45,45.46,43.87,43.26,41.09,38.86,38.40,37.74,37.02,32.76,31.85,31.15,28.58,28.33,28.12,26.51,26.46,23.63,23.40,18.72,17.44,16.94,16.45,14.35.HRMS(APCI)C40H55O6(631.4015);631.4012(M-H)-。
图2为化合物4b的高分辨质谱图。
以上实施例的反应方程式以及化合物结构式如下所示:
实施例9
将上述实施例得到的化合物进行金黄色葡萄球菌(ATCC 12228)抑菌测试,化合物的MIC数据如表1所示。甘草次酸(glycyrrhetic acid,GA,CAS号:471-53-4)对金黄色葡萄球菌(ATCC 12228)的MIC值为0.4μM。
表1化合物对金黄色葡萄球菌(ATCC 12228)的抑菌效果
实施例10
将上述实施例得到的化合物进行金黄色葡萄球菌(ATCC 6538)抑菌测试,化合物的MIC数据如表2所示。甘草次酸(glycyrrhetic acid,GA,CAS号:471-53-4)对金黄色葡萄球菌(ATCC 6538)的MIC值为0.4μM。
表2化合物对金黄色葡萄球菌(ATCC 6538)的抑菌效果
实施例11
将上述实施例得到的化合物进行金黄色葡萄球菌(ATCC 29213)抑菌测试,化合物的MIC数据如表3所示。甘草次酸(glycyrrhetic acid,GA,CAS号:471-53-4)对金黄色葡萄球菌(ATCC 29213)的MIC值为0.4μM。
表3化合物对金黄色葡萄球菌(ATCC 29213)的抑菌效果
由上述实施例及比较例可知,本发明对甘草次酸的3号碳原子和20号碳原子同时进行酯化修饰,使得其抑菌效果大大提升。
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
一种C3和C20双酯化的甘草次酸衍生物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


![一种1,3-[2H,4H]-异喹啉二酮衍生物及其制备方法和应用](https://www.zhichawang.com/images/ui/CN2019106153431/CN2019106153431.jpg)













动态评分
0.0