
]-β-环糊精HPLC柱材料的制备和用途")
IPC分类号 : B01J20/286,B01J20/29,B01J20/30,C07B57/00,C07C213/10,C07C217/10,C07C215/60,C07D215/26,C07D295/088,C07D411/04,C07D501/34,C07D501/36,C07D501/12




专利摘要
本发明涉及手性高效液相色谱柱材料的制备及应用技术领域,公开了一种β‑CD‑D2高效液相色谱手性柱材料的制备及应用,用β‑CD‑D2制备成高效液相色谱手性柱对手性药物对映异构体进行拆分。本发明内容包括:1、用β‑CD‑D2构建手性环境,与高效液相色谱硅珠键合成固定相填料,进而制备成手性柱,应用于高效液相色谱中。2、通过适当的表征手段寻找最佳配方及条件,对方法及条件优化。3、用所制得的β‑CD‑D2手性柱可建立沙美特罗单一对映体的制备方法,实现了首次用环糊精类衍生物拆分该手性化合物。4、所制得的β‑CD‑D2手性柱可拆分药物,建立了沙美特罗、特布他林、丙卡特罗、西替利嗪、拉米夫定、头孢呋辛和头孢曲松7种手性药物对映体拆分和定性定量分析方法。
权利要求
1.将双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精作为手性选择剂键合在全多孔球形硅胶基质上制备成用于高效液相色谱柱的手性固定相填料。
2.权利要求1所述的手性固定相填料,其制备方法如下:先将全多孔球形硅胶酸化,干燥后备用;将双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精在交联剂γ-(2,3-环氧丙氧基)丙基三甲氧基硅烷存在下、N2氛围中加入已处理好的全多孔球形硅胶,100℃条件下搅拌反应,反应完全后依次过滤、洗涤、干燥。
3.将权利要求1所述的手性固定相填料用于HPLC柱对沙美特罗、特布他林、丙卡特罗、西替利嗪、拉米夫定、头孢呋辛或头孢曲松进行对映体拆分。
4.将权利要求1所述的手性固定相填料用于HPLC柱对沙美特罗、特布他林、丙卡特罗、西替利嗪、拉米夫定、头孢呋辛或头孢曲松单一对映体进行定性和/或定量分析。
5.将权利要求1所述的手性固定相填料用于制备型手性HPLC柱进行沙美特罗单一对映体的制备。
说明书
技术领域
本发明涉及手性高效液相色谱(HPLC)柱材料的制备及应用技术领域,具体涉及一种双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精(β-CD-D2)HPLC手性柱材料的制备及应用,即将β-CD-D2制备成高效液相色谱(HPLC)手性柱的方法及对手性药物对映异构体的拆分研究和应用。
背景技术
高效液相色谱拆分手性药物中的光学异构体主要由试样和固定相之间的相互作用力来决定的,合成衍生出新型、耐用、高效的手性固定相是液相色谱手性拆分的重点工作之一。在拆分过程中,通过引入这些固定相来营造手性环境,通过这些固定相带来的包含作用、氢键作用、π-π共轭等实现对手性对映异构体的拆分。在HPLC手性拆分中,环糊精类手性固定相因为一些自身的优良特性得到较多重视和研究。环糊精(CD)是环状分子,具有疏水空腔和腔外亲水的羟基结构,α、β、γ-环糊精依次含6到8个葡萄糖分子。环糊精的疏水空腔能与许多含有苯、萘等疏水基团的分子发生包含作用,又因为环糊精分子上拥有多个手性中心,还能与一些含有大基团的分子有空间位阻效应,这些都使得环糊精拥有较强的手性识别能力。除此之外CD在水溶液中的溶解性较好、无毒害、对环境友好、无紫外吸收,不会对检测结果进行干扰。
在分析化学中,人们利用CD良好的选择识别能力分离各种手性物质,特别是修饰改性后的CD具有一些比母体CD更优异的性能。Xinxin Han对早期一系列环糊精衍生物作为手性固定相的手性拆分能力进行过比较。用多种环糊精的手性衍生物对30种手性呋喃的衍生物进行了手性拆分,而天然的β-CD对这些手性呋喃类化合物没有拆分效果,其中S-萘乙氨基甲酸酯基和3,5-二甲基苯基氨基甲酸酯β-CD只能对其中少部分的化合物进行拆分。近年来,不断有新的β-CD衍生物出现,Chun Lin等首先通过Staudinger反应将两种环糊精衍生物通过多个尿素与硅胶基质相连接,这两种环糊精衍生物不同之处在于一个含有甲基氨基甲酰,另一个含有氯苯基氨基甲酰,对药物的保留行为和流动相的影响做了探究,对两种不同固定相的拆分机制做了阐释。Litao Wang首次制备了β-CD-二氧化硅球型手性固定相,同时引入了乙烷基、三嗪基和3,5-二甲基苯基等多个官能团,提供了多个作用位点,还提供了类似于凝胶色谱的分子通道,能够进行非手性药物的分离,也可实现手性对映体的拆分,还有阴离子交换等特性,功能多样。最近的文献报道中,Jie Zhao用点击化学的方法制备了苯基氨基甲酰双层β-CD手性固定相。用这种双层β-CD固定相对异恶唑啉、苄氟噻嗪、吲哚洛芬、阿托品、氧化苯乙烯和丹酰基氨基酸在反相色谱模式下都实现了基线分离,其中1-(3-(4-硝基-苯基)-4,5-二氢异恶唑-5-基)吡咯烷-2-酮(4NPh-OPr)的选择系数达到5.25,分离度达到13.97。
高效液相色谱所取得的成就与固定相制备填装技术的提高密不可分,现在固定相的填充方法较为多样,主要分为密度法、粘度法、干法和匀浆法。
平衡密度法:因为硅胶的制作方法不一样,硅胶的密度有差异。需要选择与硅胶密度相近的溶剂与之混合,经常使用的是卤代烷烃,适当加入其它溶剂混合配置出密度适宜的混合液,避免基质颗粒大小不一引起的分级沉降。但是卤代烷烃的使用会造成基线难以平衡,谱图的对称性不佳,且毒性较大。
平衡粘度法:是采用诸如聚乙二醇、乙二醇、甘油之类的高粘度试剂,然后混合配制高粘度匀浆液,从而有效地阻止颗粒的沉降。但是,用这类高粘度匀浆液填充的话,压力很大,必须有更高的装柱压力,对泵的要求很高,而且装填时间很长。最后装填好的色谱柱的柱效也不高,色谱峰的对称性也差,所以很少人使用这种方法。
干法:将制备好的固定相填料依次装入空管中,同时需要震动和旋转。柱头和柱尾分别连接氮气和真空泵可以使柱效能有一定的提升,然后用溶剂高速流过色谱柱数小时。此过程要反复多次以确保色谱柱的装填没有空隙,比较适合粒径大于20μm的填料。但是干法填装还是会使填料附着在内壁上,且会因为强烈的静电作用导致相互排斥,色谱柱难以填充得均匀紧密。
匀浆法:对于粒径小于20μm的填料,会因为静电荷的作用而有很高的表面能,在干燥状态下会粘连在一起,所以对于小粒径的色谱填料需要选择一种合适的溶剂或者是几种溶剂的混合液作为分散液,常用的分散液有甲苯异丙醇(90/10)(v/v),丙酮异丙醇(50/50)(v/v),四氯化碳甲醇(95/5)(v/v),三氯甲烷甲醇(50/50)(v/v)。然后经超声处理,使填料高度分散,形成匀浆液,然后再用高压泵,用顶替液快速将填料填充到色谱柱里,得到均匀紧密的柱床。
本申请人将双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精(缩写β-CD-D2)用于高效液相色谱领域,与硅胶基质键合后,作为手性柱材料用于多种手性对映体的拆分。首先将衍生物合成后用多种分析仪器对产物结构进了表征确认,再将产物与硅胶基质相键合,对得到的β-环糊精衍生物手性固定相填料,用扫描电镜和透射电镜对其表征确认,再用匀浆法制备色谱柱。该色谱柱用于沙美特罗、西替利嗪、丙卡特罗、特布他林的手性拆分,得到了这几种β-肾上腺激动剂以及拉米夫定、头孢呋辛和头孢曲松对映体拆分的最佳高效液相色谱分离条件。
发明内容
针对现有技术中存在的不足,本发明的目的在于提供了一种新型手性高效液相色谱(HPLC)柱材料的制备及应用技术,具体涉及了一种双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精(缩写β-CD-D2)手性HPLC柱材料的制备及应用,即将β-CD-D2制备成高效液相色谱(HPLC)手性柱的方法及对手性药物对映异构体的拆分研究。这些工作是基于以下步骤进行:(1)、用β-CD-D2构建手性环境,与高效液相色谱硅珠(全多孔球形硅胶基质)键合成固定相填料,进而制备成手性柱,应用于高效液相色谱(HPLC)中。(2)、通过适当的表征手段寻找手性填料制备的最佳配方及反应。(3)用所制得的β-CD-D2手性柱可建立沙美特罗单一对映体的制备方法。(4)用所制得的β-CD-D2手性柱可拆分药物,建立了沙美特罗、特布他林、丙卡特罗、西替利嗪、拉米夫定、头孢呋辛和头孢曲松等手性药物的对映体拆分和定量分析方法,部分手性药物可通过该手性柱制备获得单一对映体。
本发明将双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精用于高效液相色谱(HPLC)领域,将其与硅胶基质键合后,作为柱材料制备手性HPLC柱,用于多种手性对映体的拆分。首先将衍生物合成后用紫外和红外对产物结构进了确认表征,对该衍生物以前未进行的荧光特性和XRD粉末衍射特性也进行了表征。将产物与硅胶基质相键合,对得到的β-环糊精衍生物手性固定相填料,用扫描电镜和透射电镜对其表征确认和筛选,再用匀浆法制备色谱柱。该色谱柱用于沙美特罗、特布他林、丙卡特罗、西替利嗪的手性拆分,得到了这几种β-肾上腺激动剂的最佳高效液相色谱分离条件,建立了相关单一对映体的制备或定量分析方法。获得了对拉米夫定、头孢呋辛和头孢曲松等多种手性物质的理想拆分效果:本发明利用双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精制成手性固定相填料,可建立多种手性物质单一对映体的HPLC拆分及定性定量检测新方法。结果显示此种制备HPLC柱的方法是有效的,可成功地将β-CD-D2键合到硅胶基质上,再填充至HPLC柱内,也进一步表明此种HPLC柱制备法是可行的。用上述β-环糊精衍生物制备手性HPLC柱的方法是有创新性的工作,在HPLC手性固定相中运用的相关工作也是有创新性的。
经中科院武汉科技查新咨询检索中心出具查新报告显示,未见文献报道。
与现有技术相比,本发明的优点和有益效果在于:
1、首次用β-环糊精衍生物作为手性固定相对沙美特罗、丙卡特罗、拉米夫定进行手性拆分,未见文献报道,该技术弥补了这一领域的空白。
2、提供了一种新型β-环糊精衍生物的高效液相色谱柱填料,确定了用双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精作为手性选择剂,制备手性HPLC柱的方法,拓展了这一物质的应用范围。
3、用双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精衍生物制成的制备型手性HPLC柱从消旋体中可得到纯化的D(+)-沙美特罗和L(-)-沙美特罗单一对映体纯品,为沙美特罗单一对映体制备提供了一种新方法。
4、在双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精衍生物手性HPLC柱上获得了可分离沙美特罗、特布他林、丙卡特罗、西替利嗪、拉米夫定、头孢呋辛和头孢曲松等手性物质的分离条件,在最佳条件下均可实现基线分离,据此可建立多种手性物质单一对映体HPLC定量测定新方法。
附图说明
图1:β-CD-D2的紫外扫描(UV)图谱;
图2a:β-CD的红外扫描(IR)图谱;
图2b:β-CD-A2的红外扫描(IR)图谱;
图2c:β-CD-D2的红外扫描(IR)图谱;
图3:β-CD,β-CD-A2,β-CD-D2的液体荧光扫描图谱;
图4:β-CD,β-CD-A2,β-CD-D2的粉末固体荧光扫描图谱;
图5a:β-CD的X射线粉末衍射(XRD)图;
图5b:β-CD-A2的X射线粉末衍射(XRD)图;
图5c:β-CD-D2的X射线粉末衍射(XRD)图;
图6a:β-CD-D2键合硅胶固定相填料在1μm时透射电镜扫描图;
图6b:裸硅胶基质在1μm时透射电镜扫描图;
图6c:β-CD-D2键合硅胶固定相填料在2μm时透射电镜扫描图;
图6d:裸硅胶基质在2μm时透射电镜扫描图;
图7a:β-CD-D2键合硅胶放大15000倍扫描电镜图;
图7b:β-CD-D2键合硅胶放大7000倍扫描电镜图;
图7c:β-CD-D2键合硅胶放大5000倍扫描电镜图;
图7d:β-CD-D2键合硅胶放大1500倍扫描电镜图;
图7e:裸硅胶基质放大25000倍扫描电镜图;
图7f:裸硅胶基质放大10000倍扫描电镜图;
图7g:裸硅胶基质放大1300倍扫描电镜图;
图7h:板结的β-CD-D2键合硅胶放大9000倍扫描电镜图;
图7l:板结的β-CD-D2键合硅胶放大4000倍扫描电镜图;
图8:由β-CD-D2键合硅胶HPLC手性柱拆分沙美特罗标准品与溶剂的对照实验;
图9:由β-CD-D2键合硅胶HPLC手性柱制备沙美特罗两对映异构体HPLC图;
图10:由β-CD-D2键合硅胶HPLC手性柱制备所得左旋沙美特罗单一对映体的圆二色谱图;
图11a:特布他林拆分HPLC图;
图11b:特布他林拆分溶剂对比HPLC图;
图12a:丙卡特罗拆分HPLC图;
图12b:丙卡特罗拆分溶剂对比HPLC图;
图13a:拉米夫定拆分HPLC图;
图13b:拉米夫定拆分溶剂对比HPLC图
图14:西替利嗪拆分HPLC图;
图15:头孢呋辛拆分HPLC图;
图16:头孢曲松拆分HPLC图。
具体实施方式
下面申请人将结合具体的实施例对本发明的新手性HPLC柱的制备和应用过程做详细说明,目的在于使本领域技术人员对本发明有更进一步的理解。
实施例1:双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精(β-CD-D2)的制备
依据文献(丁志刚,任维衡,习玲玲等,β-环糊精衍生物的合成及对脲酶的抑制作用.催化化学,1996,6(17):567-569.)制得β-CD-D2后,用紫外、红外光谱、荧光和XRD等手段确定是所要目标化合物。
具体做法:先将马来酸酐和β-CD(摩尔比15:1)置于恒温水浴80℃反应8小时,停止反应,后处理得粉末状产物即为β-CD-马来酸酐(简称β-CD-A2),置于干燥器中备用。将β-CD-A2和氯乙酸(摩尔比1:4)置于锥形瓶中,在80℃恒温水浴锅中反应8小时,取出冷却至室温,后处理得白色粉末状产物即为β-CD-D2。
具体反应过程如下所示:
1、紫外扫描(UV)图谱
图1中的所有化合物均以超纯水为溶剂,浓度为1.0×10-6mol/L。a为β-CD,在近紫外光区没有吸收,是因为β-CD只有碳-碳单键,碳-氧单键,有不饱和键,发生的是σ-σ*,n-σ的跃迁,需要较高能量,吸收波长在远紫外光区。b为β-CD-A2,因为马来酸酐的引入,有了碳碳双键,碳氧双键,且有π-π共轭效应,所以在230nm处有强吸收。c为β-CD-D2,其中的碳碳双键被破坏,π-π共轭效应消失,所以210nm处出现酯基和羧基的特征弱吸收。紫外光谱的谱图初步说明得到了目标产物。
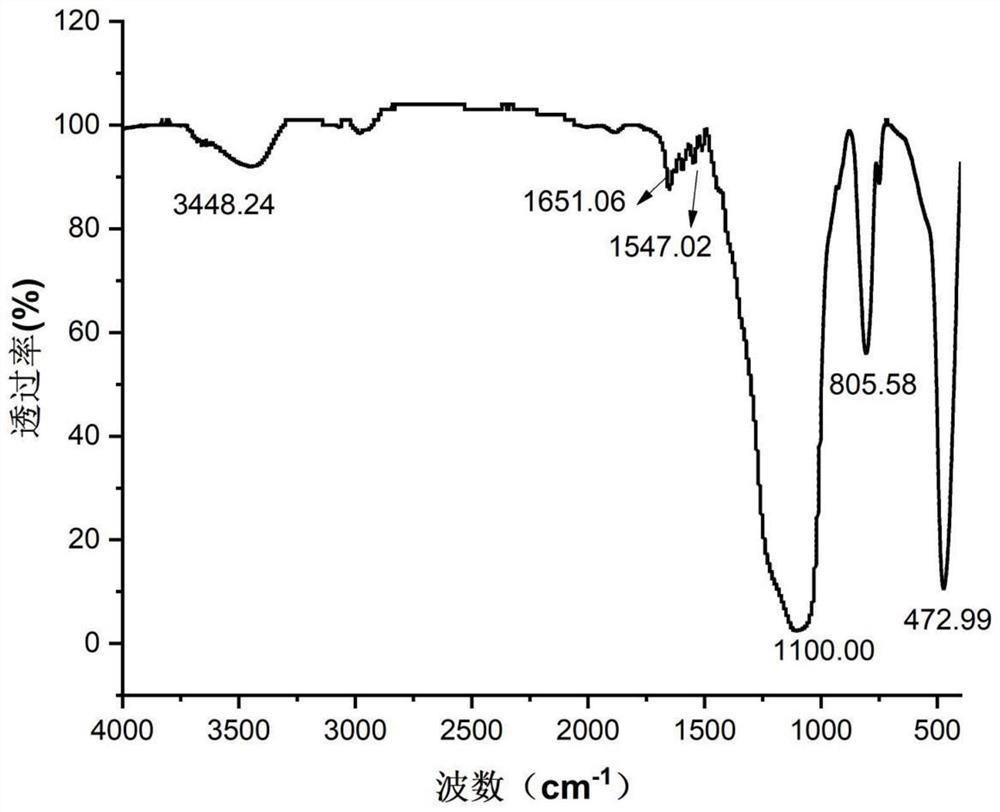
2、红外扫描(IR)图谱
图2b(β-CD-A2的IR图)与图2a(β-CD的IR图)相比较明显多出了1725cm-1处的C=O的伸缩振动峰,1640cm-1处的C=C的伸缩振动峰,且C=O与C=C共轭,谱带强度增大。1157cm-1,1220cm-1处有C-O-C的伸缩振动谱带,说明马来酸酐接在了β-环糊精上。图2c(β-CD-D2的IR图)与图2b相比较,依然有1725cm-1处的C=O的伸缩振动峰,但是因为没有C=C,没法形成共轭,谱带强度明显减弱,进一步说明最终产物合成成功。
3、荧光光谱
图3中a、b、c分别为β-CD、β-CD-A2、β-CD-D2对应的液体荧光扫描图谱,β-CD、β-CD-A2、β-CD-D2都以超纯水为溶剂,浓度均为1.0×10-6mol/L。可看出在水溶液中3种物质结构上的共轭差异无法体现出来,所得最大荧光发射波长峰都在274nm附近,说明溶液中主要以β-CD环产生的发射波长为主导。
图4中a、b、c分别为β-CD、β-CD-A2、β-CD-D2对应的固体荧光扫描图谱;固体粉末状时三种结构的物质产生的共轭体系有差异,故而能看出在298nm激发波长的照射下得出了不同的发射波长扫描图。
4、X射线粉末衍射(XRD)图
图谱5b较谱图5a的锐利衍射峰明显增加,响应增强,这可能是因为马来酸酐引入后,双键使β-CD环糊精的刚性结构增强,分子更加有序。当双键破坏以后,图谱5c中的锐利衍射峰减少。说明β-CD、中间产物β-CD-A2以及最终产物β-CD-D2的结构有一定差异,进一步说明产物合成成功。
实施例2β-CD-D2手性HPLC柱的制备
用实施例1合成的手性分离材料——双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精(β-CD-D2)作为HPLC手性选择剂,制备手性HPLC柱填料,进而制备高效液相色谱HPLC柱,用透射电镜和扫描电镜,可确认填料制备的优劣。
1、高效液相色谱柱填料的制备方法
制备方法为:先将商用粒径5μm的硅珠(全多孔球形硅胶基质)酸化,干燥后备用;β-CD-D2在交联剂γ-(2,3-环氧丙氧基)丙基三甲氧基硅烷(β-CD-D2与交联剂摩尔比5:1)存在下、N2氛围中,加入已处理好的全多孔球形硅胶(本实施例β-CD-D2与全多孔球形硅胶质量比为1:2),100℃条件下搅拌反应24h,反应完全后过滤、洗涤、干燥,备用。
2、手性HPLC柱填料的表征
把制备好的手性HPLC柱填料先用透射电子显微镜观察显示明显较未反应的裸硅胶基质颗粒大,再用场发射扫描电镜(ZEISS SIGMA)扫描表征,扫描结果如图6、图7所示。
从图6可见:a、c分别是β-CD-D2固定相填料颗粒在透射电镜放大到1μm、2μm时的表征图谱,b、d为裸硅胶基质在透射电镜放大到1μm、2μm时的图谱,可以看出键合β-CD-D2后的硅胶直径比未键合的硅胶明显增大。
由图7可知,a、b、c、d分别是β-CD-D2键合硅胶放大15000倍、7000倍、5000倍、1500倍的扫描电镜谱图,e、f、g是裸硅珠放大25000倍、10000倍、1300倍的扫描电镜谱图,可以清楚看到裸硅胶基质表面光滑(如e、f、g所示),即使放大25000倍也是光滑的,而键合后的硅胶基质被β-CD-D2均匀包裹,理想键合反应后硅胶表面呈现出均匀的层状物质,与反应条件控制不佳的h、l(β-CD-D2与全多孔球形硅胶质量比1.7:1)中发生板结情况的硅胶基质完全不同,这也是保证高柱效的前提。
3、固定相的装填
采用匀浆法进行装填,高压泵一次填装进各种规格液相色谱不锈钢空柱当中,冲洗,甲醇饱和。
通过UV、IR、荧光和XRD对β-CD-D2的合成进行了表征,用TEM,SEM对β-CD-D2HPLC固定相填料键合前后的硅胶基质进行了表征,达到预期目标。用匀浆法制得了β-CD-D2手性固定相高效液相色谱柱。
4、所用的仪器与试剂分别为:
PE Lambda Bio35紫外-可见分光光度计(美国);NEXUS 470型智能傅立叶红外光谱仪(美国Thermo Nicolet公司);VG Multilab 2000光电子能谱仪(美国);Tecnai G220S-TWIN透射电子显微镜(捷克);Hitachi SU8010型扫描电子显微镜(日本);酸度计(上海伟业,pHs-3c型);β-环糊精(国药集团);顺丁烯二酸酐(国药集团);氯乙酸(天津市亚丽区天大化学试剂);全多孔球形硅胶(兰州化物所)。
实施例3用实施例2制备的手性双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精手性HPLC柱分离手性药物
将双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精制备成β-CD-D2手性固定相填料,进而装填为高效液相色谱柱用于手性物质的拆分,可建立分离沙美特罗、特布他林、丙卡特罗、拉米夫定、西替利嗪、头孢呋辛和头孢曲松7种手性药物的对映体拆分和定量分析方法,还可通过该手性柱制备获得沙美特罗单一对映体,据此可建立多种手性物质单一对映体HPLC定量测定新方法。
1、主要仪器及试剂
Thermo UltiMate 3000(赛默飞世尔科技(中国)有限公司),LC-15C岛津高效液相色谱仪(岛津企业管理(中国)有限公司),依利特液相色谱仪(分析型P230Ⅱ,制备型P270);紫外检测器(分析型UV230,Ⅱ制备型UV230+),As3120超声波清洗器(宁波科生仪器厂),ZD-2型酸度计(上海伟业仪器厂),CJ-1型电磁搅拌器(上海华光仪器仪表厂),溶剂过滤器(上海津腾有限公司),0.2μm微孔滤膜(上海市新亚净化器件厂),100μL微量进样器(岛津企业管理(中国)有限公司),BP211D电子分析天平(sartorius),乙腈(AR,国药集团化学试剂有限公司),磷酸二氢钠(国药集团化学试剂有限公司),三乙胺(国药集团化学试剂有限公司),超纯水(美国Moleculer超纯水机生产),沙美特罗替卡松粉吸入剂(葛兰素史克公司),沙美特罗标准品(中国食品药品检定研究院)特布他林标准品(中国食品药品检定研究院),丙卡特罗标准品(中国食品药品检定研究院)。拉米夫定对映体混合物(中国食品药品检定研究所),西替利嗪(中国药检所)。
市售舒利迭沙美特罗替卡松粉吸入剂每吸(泡)内含50微克沙美特罗(昔萘酸盐形式)和250微克丙酸氟替卡松。用甲醇溶剂制得沙美特罗溶液样品1.1×10-4mol/L作为原液,实验过程中根据需要稀释到不同浓度进行使用。每次进样前均由0.22μm的有机系滤膜过滤。
标准样品为昔萘酸沙美特罗化学对照品,用甲醇溶剂制得沙美特罗标准品溶液1.3×10-3mol/L作为原液,实验过程中根据需要稀释到不同浓度进行使用。每次进样前均由0.22μm的有机系滤膜过滤。
2、沙美特罗的分离
采用按照实施例2制备的β-CD-D2手性HPLC柱(柱尺寸150mm×Φ4.6mm),用HPLC仪对沙美特罗进行了手性拆分,最佳条件为:缓冲液磷酸二氢钠-三乙胺和乙腈为流动相,缓冲液pH值9.2,缓冲液浓度为0.5mmol/L,V(缓冲液):V(乙腈)=65:35,沙美特罗浓度1.1×10-4mol/L,流速0.5mL/min,进样量20uL,检测波长254nm,温度20。℃分离度Rs为2.88,如图8a所示,实现了基线分离,沙美特罗浓度在1.325×10-5mol/L~2.650×10-4mol/L范围左旋和右旋沙美特罗两对映体与峰高和峰面积均有线性相关性,前峰峰高-浓度标准曲线方程为Y=0.08469X-0.3713(r=0.9901),前峰峰面积-浓度标准曲线方程为Y=15.005X-71.46(r=0.9995);后峰峰高-浓度标准曲线方程为Y=0.9979+2.726X(r=0.9956),后峰峰面积-浓度标准曲线方程为Y=1.031×103X-3.550×102(r=0.9997)。重复进样7次,两峰峰高峰面积RSD均在10%以内。经验证溶剂甲醇对实验无影响,如图8b所示。沙美特罗两对映体洗脱强度大,峰形对称,出峰时间短,本实验建立了一种新的沙美特罗两对映体的拆分检测方法,在图9条件(流动相体积比为V(缓冲液):V(乙腈)=85:15,其余条件同前。)下制备(柱尺寸150mm×Φ10mm,将该柱用于进样体积为10mL的制备型HPLC仪。)收集前后峰与溶剂的混合溶液测定圆二色谱,确定后峰为左旋沙美特罗单一对映体,如图10所示。
3、特布他林的分离
特布他林(1.98×10-3mol/L,溶剂为超纯水,进样体积20μL)在β-CD-D2制备的手性柱(柱尺寸150mm×Φ4.6mm)上拆分最佳条件为,缓冲液柠檬酸-三乙胺,pH值为3.5,柠檬酸浓度26.1mmol/L,流动相由缓冲液柠檬酸-三乙胺、四氢呋喃和乙腈按照65:10:25(v:v:v)的比例组成,流速0.5mL/min,柱温25。℃分离度Rs达到3.51,实现了基线分离,如图11a所示,确定了溶剂峰无干扰,如图11b所示。且重复进样7次,各组分的相关参数RSD均在6.24%以内,特布他林消旋体浓度在9.9×10-5mol/L-3.96×10-4mol/L的浓度范围内,具有线性相关性,前峰峰高-浓度标准曲线方程为y=0.7577+10.978x,线性相关系数r=0.9976,前峰峰面积-浓度标准曲线方程为y=-0.01257+0.5811x,线性相关系数r=0.9970;后峰峰高-浓度标准曲线方程为y=2.846+3.304x,线性相关系数r=0.9808,后峰峰面积-浓度标准曲线方程为y=0.07912+0.3099x,线性相关系数r=0.9948。用峰面积定量,特布他林两对映体的线性相关系数可达到0.99以上。
4、丙卡特罗的分离
用β-CD-D2手性HPLC柱(柱尺寸150mm×Φ4.6mm)对丙卡特罗标准品(1.70×10-4mol/L,溶剂为超纯水,进样体积20μL),在反相模式下进行了手性拆分,成功分离了丙卡特罗(R,R/S)和(S,R/S)光学异构体,并对分离条件进行了优化,最大分离度RS达到6.61(流动相配比V甲醇:V异丙醇:V缓冲液=80:15:5,缓冲液pH值为8.7,磷酸二氢钠-三乙胺缓冲液中磷酸二氢钠浓度为5mmol/L,温度为20,℃检测波长为280nm,流动相流速1.2mL/min),如图12a所示。重复进样9次,两对映体即前后峰的各项色谱参数RSD均在6.45%以下,丙卡特罗标准品在3.51×10-5mol/L到1.79×10-3mol/L浓度范围内前后峰峰高、峰面积与浓度有线性相关性,前峰峰高-浓度标准曲线方程为y=-5.6887+16.6883x(r=0.9914),前峰峰面积-浓度标准曲线方程为y=-24.7230+25.4066x(r=0.9977),后峰峰高-浓度标准曲线方程为y=-5.6887+16.6883x(r=0.9934),后峰峰面积-浓度标准曲线方程为y=-24.7230+25.4066x(r=0.9977)。(两对映体的线性相关系数均达到0.99以上),而且确定了溶剂峰的干扰是可以扣除的(如图12b所示)。
5、拉米夫定的分离
用β-CD-D2手性HPLC柱(柱尺寸150mm×Φ4.6mm)对拉米夫定进行拆分,用缓冲液柠檬酸三乙胺和乙腈组成流动相,缓冲液柠檬酸三乙胺浓度为5.0×10-3mol/L且pH为4.0,柠檬酸三乙胺与乙腈的体积比为45:55;拉米夫定浓度为2.26×10-4mol/L(溶剂为超纯水),进样量为20μL;流动相流速为0.8mL/min,紫外检测波长为270nm,温度为25℃,成功实现了拉米夫定的最佳分离,分离度为4.35,拉米夫定的分离结果如图13a所示。拉米夫定对映体混合物在1.0×10-5~9.04×10-4mol/L浓度范围内,样品浓度与峰高、峰面积有线性相关性,对映体前峰的峰高与浓度的线性方程为y=2.218+24.784x,相关系数r=0.9886,对映体前峰的峰面积与浓度的线性方程为y=520.564+891.062x,相关系数r=0.9716,对映体后峰的峰高与浓度的线性方程为y=-1.777+6.458x,相关系数r=0.9900,对映体后峰的峰面积与浓度的线性方程为y=105.226+218.568x,相关系数r=0.9749。(用峰高定量,两对映体的线性相关系数可达到0.99),在最佳分离条件下,重复进样8次,前后峰峰高、峰面积和半高宽的RSD均在8.17%以内。而且确定了溶剂峰的干扰是可以扣除的(如图13b所示)。
6、西替利嗪的分离
用β-CD-D2手性HPLC柱(柱尺寸150mm×Φ4.6mm)对西替利嗪(1.20×10-4mol/L,溶剂为超纯水,进样量为20μL)进行拆分,在流动相配比V(缓冲液):V(甲醇)=85:15,缓冲液pH值为7.5,缓冲液磷酸二氢钠-三乙胺中磷酸二氢钠浓度为5mmol/L,温度为25,℃检测波长为270nm,流动相流速1.0mL/min的最佳条件下,分离结果如图14所示。
7、头孢呋辛和头孢曲松的分离
分别用β-CD-D2手性HPLC柱(柱尺寸150mm×Φ4.6mm)对头孢呋辛和头孢曲松进行拆分(2.17×10-3mol/L,溶剂为超纯水,进样体积20μL),在流动相配比V(缓冲液):V(乙腈)=85:15,缓冲液pH值为3.0,缓冲液柠檬酸-三乙胺中柠檬酸浓度为5mmol/L,温度为25,℃检测波长为270nm,流动相流速1.0mL/min的最佳条件下,对头孢呋辛和头孢曲松的分离结果如图15和图16所示。
双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精HPLC柱材料的制备和用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。








![双[-6-氧-(3-脱氧柠檬酸单酯-4)]-β-环糊精HPLC柱材料的制备和用途](https://www.zhichawang.com/images/ui/CN2017102109394/CN2017102109394.jpg)









动态评分
0.0