









专利摘要
本发明公开了一种氮桥连三芳氧基稀土金属化合物及其制备方法和其催化应用,该稀土金属化合物的通式为:LLn(THF)3,其化学结构式如下:其中,通式中L表示氮桥连三芳氧基配体,配体LH3的通式为2,2',2″-[Nitrilotris(methylene)]tris[4-R1-6-R2-phenol];Ln为稀土金属,选自镧、钕、钐或钇中的一种;R1和R2选自CH3、But、Cl中的一种。本发明同时提供了上述化合物的制备方法及将其作为催化剂催化环氧烷和异氰酸酯环加成反应的应用方法。本发明的稀土金属化合物合成简单,结构明确,且收率高,应用方法条件温和,活性高,选择性好,底物适应范围广。
权利要求
1.一种氮桥连三芳氧基稀土金属化合物,其特征在于,其通式为LLn(THF)3,其化学结构式如式I:
式I
其中:通式中L表示氮桥连三芳氧基配体,其化学结构式如式II;Ln为稀土金属,选自镧、钕、钐或钇中的一种;R1和R2选自CH3、But、Cl中的一种
式II。
2.一种权利要求1所述氮桥连三芳氧基稀土金属化合物的制备方法,其特征在于,包括以下步骤:
(1)合成配体LH3
其中,LH3为2,2',2″-[Nitrilotris(methylene)]tris[4-R1-6-R2-phenol,R1和R2选自CH3、But、Cl中的一种;
(2)合成氮桥连三芳氧基稀土金属化合物
在一除水除氧、惰性气体保护的反应容器中加入LnCp3(THF),加入醚类溶剂,在另一个除水除氧、惰性气体保护的反应容器中称取与LnCp3(THF)等摩尔比的配体LH3,用醚类溶剂溶解,将配体溶液缓慢加入到LnCp3(THF)醚类溶剂的悬浊液中,溶液逐渐变澄清,反应12~24小时,最终得到澄清液;
(3)除去溶剂,再加入四氢呋喃溶解固体,离心,取上层清液,室温静置,析出晶体,即为氮桥连三芳氧基稀土金属化合物LLn(THF)3;
其中,Ln为稀土金属,选自镧、钕、钐、或钇中的一种。
3.根据权利要求2所述的一种氮桥连三芳氧基稀土金属化合物的制备方法,其特征在于,所述步骤(1)中配体LH3合成方法为下述中的一种:
(1)按40:3.0~3.3:14.0~14.2的摩尔比分别取2,4-二甲基苯酚、六次甲基四胺和30~40%的甲醛溶液,放入反应容器中反应,36~48小时后终止反应,用去离子水洗2~4次,之后用无水MgSO4、无水Na2SO4或者无水CaCl2干燥剂干燥,然后浓缩,得到油状物,用柱层析法分离,得到产物2,2,2-[Nitrilotris(methylene)]tris[4,6-dimethylphenol];
(2)按40:3.0~3.3:14.0~14.2的摩尔比取2,4-二氯苯酚、六次甲基四胺和30~40%的甲醛溶液,放入反应容器中反应,3~6小时后终止反应,用去离子水洗2~4次,之后用无水MgSO4、无水Na2SO4或者无水CaCl2干燥剂干燥,然后浓缩,得到油状物,用柱层析法分离,得到产物2,2’,2”-[Nitrilotris(methylene)]tris[4,6-dichlorophenol];
(3)按40:3.0~3.3:14.0~14.2的摩尔比取2-叔丁基对甲苯酚、六次甲基四胺和30~40%的甲醛溶液,放入反应容器中反应,36~48小时后终止反应,用去离子水洗2~4次,之后用无水MgSO4、无水Na2SO4或者无水CaCl2干燥剂干燥,然后浓缩,得到油状物,用柱层析法分离,得到产物2,2',2″-[Nitrilotris(methylene)]tris[6-tert-butyl-4-methylphenol]。
4.根据权利要求3所述的一种氮桥连三芳氧基稀土金属化合物的制备方法,其特征在于,所述配体LH3合成方法中反应温度为120℃~140℃;反应用氯仿终止;所述柱层析法分离时,先用乙酸乙酯:石油醚=1:50~1:100的洗脱剂淋洗,洗去杂质,再用乙酸乙酯:石油醚=1:10~1:20的洗脱剂淋洗。
5.根据权利要求2所述的一种氮桥连三芳氧基稀土金属化合物的制备方法,其特征在于,所述步骤(2)中的醚类溶剂为四氢呋喃、乙醚、乙二醇二甲醚中的一种。
6.一种如权利要求1所述的氮桥连三芳氧基稀土金属化合物作为催化剂催化环氧烷和异氰酸酯环加成反应的应用。
7.根据权利要求4所述的氮桥连三芳氧基稀土金属化合物作为催化剂催化环氧烷和异氰酸酯环加成反应的应用,其特征在于,该应用的方法包括以下步骤:
(1)在无水无氧、惰性气体保护下,将氮桥连三芳氧基稀土金属化合物以及添加剂加入反应容器中,加入溶剂,再加入环氧化合物和异氰酸酯,置于设定反应温度恒温搅拌,进行反应;
(2)步骤(1)中的反应结束后,原位法取出一滴,放入核磁管中,加入氘代氯仿,进行核磁表征,计算反应产率,剩余物用乙酸乙酯萃取,再经柱层析分离,用乙酸乙酯:石油醚=1:2~1:10的洗脱剂淋洗,得到产物。
8.根据权利要求7所述的氮桥连三芳氧基稀土金属化合物作为催化剂催化环氧烷和异氰酸酯环加成反应的应用,其特征在于,步骤(1)中,所述环氧烷、异氰酸酯和催化剂的摩尔比为50~300:50~300:1;反应温度为60~120℃;反应时间为12~24小时。
9.根据权利要求7所述的氮桥连三芳氧基稀土金属化合物作为催化剂催化环氧烷和异氰酸酯环加成反应的应用,其特征在于,步骤(1)中,所述溶剂选自己烷、甲苯、苯、二甲苯、溴苯、二氯甲烷、环碳酸酯或二氯乙烷中的一种。
10.根据权利要求7所述的氮桥连三芳氧基稀土金属化合物作为催化剂催化环氧烷和异氰酸酯环加成反应的应用,其特征在于,步骤(1)中,所述添加剂为四丁基碘化铵,四丁基溴化铵,四丁基氯化铵,四辛基溴化铵,双(三苯基膦)氯化铵或者苄基三丁基溴化铵中的一种。
说明书
技术领域
本发明涉及一种催化剂的制备技术领域,具体涉及一种氮桥连三芳氧基稀土金属化合物及其制备方法和在催化环氧烷和异氰酸酯环加成反应中的应用。
背景技术
恶唑烷酮类化合物是一种重要的有机合成中间体,具有广泛的用途。比如恶唑烷酮类化合物可以用来合成一些抗细菌(如:分支杆菌,葡萄杆菌,等)的药物等。因此,近年来恶唑烷酮类化合物受到了人们越来越多的关注。合成恶唑烷酮类化合物的一种主要方法就是环氧烷与异氰酸酯的偶合加成反应。目前,已发现一些催化剂对此类反应有较好的催化效果,主要包括鎓盐类催化体系和金属化合物催化体系两种。
关于鎓盐催化体系的报道:
1958年,Speranza,G.P.和Peppel,W.J.报道了用TEAB催化异氰酸酯和环氧烷的环加成反应,但该反应需要在160℃以上的温度下才能得到较高的产率。(参见:Speranza,G.P.;Peppel,W.J.J.Org.Chem.1958,23,1922-1924.)。
关于金属化合物催化体系的报道:
(1)1986年,Shibata,I.课题组用烷基锡卤化物和碱组成的催化体系催化异氰酸酯和环氧烷的环加成反应,在40℃下,反应2个小时能达到较高的收率。但是该反应需要的催化剂用量很高,是环氧烷类化合物用量的10%,且异氰酸酯用量是环氧烷的五倍。(参见:Shibata,I.;Baba,A.;Iwasaki,H.;Matsuda,H.J.Org.Chem.1986,51,2177-2184.)。
(2)1990年Baba,A.课题组用四苯基碘化锑催化异氰酸酯和环氧烷的环加成反应,在40℃下,反应一个小时可以当量转化。但是需要的催化剂量很高,用量是异氰酸酯的10%,并且区域选择性也不高。(参见:Fujiwara,M.;Baba,A.;Matsuda,H.Bull.Chem.Soc.Jpn.1990,63,1069-1073)。
(3)2010年,Barros,M.T.和Phillips,A.M.F.等以稀土或过渡金属氯化物催化异氰酸酯和环氧烷的不对称环加成反应。以20mol%的催化剂用量,可以得到84%的产率以及75%的e.e.值。(参见:Barros,M.T.;Phillips,A.M.F.Tetrahedron:Asymmetry.2010,21,2746–2752)。
(4)2013年,North,M.课题组报导了双Salen铝化合物可以催化异氰酸酯和环氧烷的环加成反应,用5%的催化剂用量,在80℃下,反应18小时,可以当量转化,并且有较好的区域选择性。(参见:Baronsky,T.;Beattie,C.;Harrington,R.W.;Irfan,R.;North,M.;Osende,J.G.,Young,C.ACS Catal.2013,3,790-797)。
尽管这些催化体系都能催化异氰酸酯和环氧烷的环加成反应,得到恶唑烷酮类化合物,但是在这些体系中存在着诸多问题,如:催化剂的区域选择性差、催化剂的用量大、反应条件苛刻、底物的普适性差等等。因此,寻找一种原料来源简单、反应条件温和、普适性好的制备方法以高效地合成恶唑烷酮类化合物是有意义的。
发明内容
本发明目的是克服现有技术的不足,提供一种氮桥连三芳氧基稀土金属化合物,本发明中的氮桥连三芳氧基稀土金属化合物可以作为催化剂催化环氧烷和异氰酸酯的环加成反应,制备恶唑烷酮类化合物,其催化活性高,区域选择性好,底物适应范围广。
本发明的技术方案是:
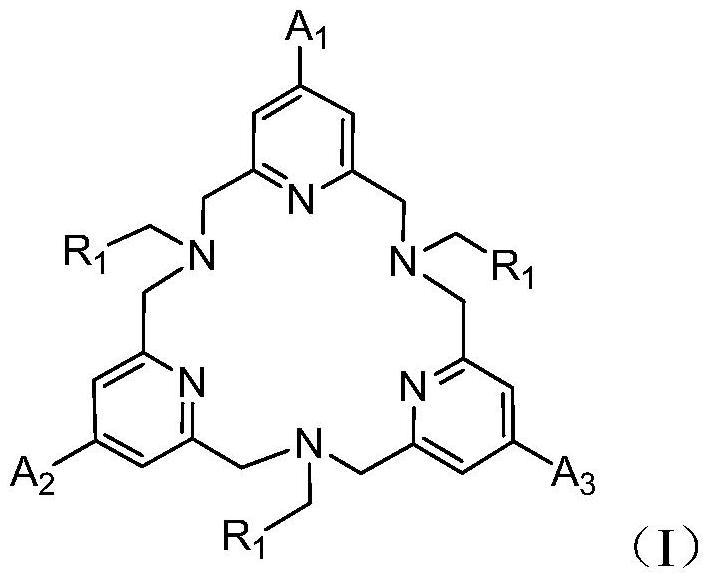
一种氮桥连三芳氧基稀土金属化合物,其通式为LLn(THF)3,其化学结构式如下式I:
式I
其中:通式中L表示氮桥连三芳氧基配体,其化学结构式如式II;Ln为稀土金属,选自镧、钕、钐、或钇中的一种;R1和R2选自CH3、But、Cl中的一种,
式II。
本发明同时提供一种所述氮桥连三芳氧基稀土金属化合物的制备方法,
该制备方法中涉及配体的合成,该配体的合成为现有技术其方法可参考文献(1).Dargaville,T.R.;Bruyn,P.J.D.;Lim,A.S.C.;Looney,M.G.;Potter,A.C.;Solomon,D.H.;Zhang,X.Q.Chemistry of Novolac Resins 1997,1389-1398.(2).Groysman,S.;Goldberg,I.;Kol,M.;Genizi,E.;Goldschmidtb,Z.Adv.Synth.Catal 2005,347,409–415.(3).Timosheva,N.V.;Chandrasekaran,A.;Day,R.O.;Holmes,R.R.Organometallics 2001,20,2331-2337.
所述的氮桥连三芳氧基稀土金属化合物的制备方法具体包括以下步骤:
(1)合成配体LH3:
LH3通式为2,2',2″-[Nitrilotris(methylene)]tris[4-R1-6-R2-phenol,其中R1和R2选自CH3、But、Cl中的一种;
(2)合成氮桥连三芳氧基稀土金属化合物
在一除水除氧、惰性气体保护的反应容器中加入LnCp3(THF),加入醚类溶剂,在另一个除水除氧、惰性气体保护的反应容器中称取与LnCp3(THF)等摩尔比的配体LH3,用醚类溶剂溶解,将配体溶液缓慢加入到LnCp3(THF)醚类溶剂的悬浊液中,溶液逐渐变澄清,反应12~24小时,最终得到澄清液;
(3)除去溶剂,再加入四氢呋喃溶解固体,离心,取上层清液,室温静置,析出晶体,即为氮桥连三芳氧基稀土金属化合物LLn(THF)3;其反应方程式如下:
其中,Ln为稀土金属,选自镧、钕、钐、或钇中的一种。
进一步的,步骤(2)中所述醚类溶剂为四氢呋喃、乙醚或乙二醇二甲醚中的一种。
进一步的,所述步骤(1)中配体LH3合成方法为下述中的一种:
(1)2,2,2-[Nitrilotris(methylene)]tris[4,6-dimethylphenol]的合成:按40:3.0~3.3:14.0~14.2的摩尔比分别取2,4-二甲基苯酚、六次甲基四胺和30~40%的甲醛溶液,放入反应容器中反应,36~48小时后终止反应,用去离子水洗2-4次,之后用无水MgSO4或者无水NaSO4或者无水CaCl2等干燥剂干燥,然后浓缩,得到油状物,用柱层析法分离,得到产物。其反应方程式如下:
(2)2,2’,2”-[Nitrilotris(methylene)]tris[4,6-dichlorophenol]的合成:按40:3.0~3.3:14.0~14.2的摩尔比取2,4-二氯苯酚、六次甲基四胺和30~40%的甲醛溶液,放入反应容器中反应,3~6小时后终止反应,用去离子水洗2-4次,之后用无水MgSO4、无水NaSO4或者无水CaCl2等干燥剂干燥,然后浓缩,得到油状物,用柱层析法分离,得到产物。其反应方程式如下:
(3)2,2',2″-[Nitrilotris(methylene)]tris[6-tert-butyl-4-methylphenol]的合成:按40:3.0~3.3:14.0~14.2的摩尔比取2-叔丁基对甲苯酚、六次甲基四胺和30~40%的甲醛溶液,放入反应容器中反应,36~48小时后终止反应,用去离子水洗2-4次,之后用无水MgSO4或者无水NaSO4或者无水CaCl2等干燥剂干燥,然后浓缩,得到油状物,用柱层析法分离,得到产物。其反应方程式如下:
进一步的,配体LH3合成方法中所述反应温度为120~140℃;
进一步的,配体LH3合成方法中所述反应用氯仿终止反应;
进一步的,配体LH3合成方法中所述柱层析法分离时,先用乙酸乙酯:石油醚=1:50~1:100的洗脱剂淋洗,洗去杂质,再用乙酸乙酯:石油醚=1:10~1:20的洗脱剂淋洗,得到产物。
本发明还提供了上述制得的氮桥连三芳氧基稀土金属化合物作为催化剂催化环氧烷和异氰酸酯环加成反应中的应用。
其应用方法包括如下步骤:
(1)在无水无氧、惰性气体保护下,将氮桥连三芳氧基稀土金属化合物以及添加剂加入溶剂中,再加入环氧化合物和异氰酸酯,置于设定反应温度的恒温浴中搅拌,进行反应;
(2)步骤(1)中的反应结束后,原位法取出一滴,放入核磁管中,加入氘代氯仿,进行核磁表征,计算反应产率,剩余物用乙酸乙酯萃取,再经柱层析分离,用乙酸乙酯:石油醚=1:2~1:10的洗脱剂淋洗,得到产物。
步骤(1)和(2)合成工艺的反应方程式如下:
其中R1、R2、R3独立地选自烷基、芳基、卤素、酯基、醚基以及羟基中的一种。
进一步,优选的,上述应用方法步骤(1)中,环氧化物、异氰酸酯和催化剂的摩尔比为50~300:50~300:1;
进一步,优选的,上述应用方法步骤(1)中,反应温度为60~120℃;
进一步,优选的,上述应用方法步骤(1)中,反应时间为12-24小时;
进一步,优选的,上述应用方法步骤(1)中,所述的溶剂选自己烷、甲苯、苯、二甲苯、二氯甲烷、溴苯、环碳酸酯或二氯乙烷中的一种。
更进一步,优选的,上述应用方法步骤(1)中,所述添加剂为四丁基碘化铵,四丁基溴化铵,四丁基氯化铵,四辛基溴化铵,双(三苯基膦)氯化铵或者苄基三丁基溴化铵中的一种。
本发明与现有技术相比具有下列优点:
1.本发明利用的氮桥连三芳氧基稀土金属化合物结构明确,合成方法简单,产率高,分离纯化简单;添加剂季铵盐的来源广泛。
2.本发明公开的催化剂活性高,其催化剂的用量为反应物环氧烷的0.5mol%,添加剂的用量为反应物环氧烷的1mol%时,产物收率大于95%,较少的催化剂用量也有利于产物的提纯。
3.本发明公开的制备方法中原料易得、反应条件温和、反应底物普适性广,不仅能高效催化单取代环氧烷与异氰酸酯反应,还适用于双取代环氧烷与异氰酸酯的反应;反应时间短,目标产物的收率高,反应操作过程简单。
具体实施方式
下面结合具体实施例对本发明的技术方案进一步详细说明。
本发明的核心是将氮桥连三芳氧基用作配体制备氮桥连三芳氧基稀土金属化合物,该化合物的通式为:LLn(THF)3。
实施例1:制备L1La(THF)3
其步骤如下:
(1)合成配体L1H3,其制备方法如下:
2,2,2-[Nitrilotris(methylene)]tris[4,6-dimethylphenol](L1H3)的合成:按40:3.3:14.2的摩尔比分别取2,4-二甲基苯酚、六次甲基四胺和35%的甲醛溶液,放入圆底烧瓶中反应,在130℃下反应48小时后,降至室温,氯仿终止反应,用去离子水洗3次,之后用MgSO4干燥,然后浓缩,得到油状物,最后柱层析。先用乙酸乙酯:石油醚=1:50的洗脱剂淋洗,洗去杂质,再用乙酸乙酯:石油醚=1:10的洗脱剂淋洗,浓缩,析出白色固体,抽干,得到化合物L1H3,产率40%。核磁数据:1H NMR(400MHz,CDCl3):6.90-6.85(s,3H,ArH);6.75-6.72(s,3H,ArH);3.75-3.5(s,6H,CH2);2.30-2.10(s,18H,CH3)。
(2)合成氮桥连三芳氧基稀土金属化合物L1La(THF)3
在一除水除氧、氩气保护的反应瓶中加入LaCp3(THF),加入四氢呋喃溶剂,在另一个除水除氧、氩气保护的反应瓶中称取与LaCp3(THF)等摩尔比的配体L1H3,用四氢呋喃溶剂溶解,将配体溶液缓慢加入到LaCp3(THF)的四氢呋喃悬浊液中,溶液逐渐变澄清,反应24小时,最终得到澄清液。
(3)抽干溶剂,再加入四氢呋喃至恰好溶解,离心,取上层清液至结晶瓶中,室温静置直至析出晶体,即为氮桥连三芳氧基稀土金属化合物L1La(THF)3。
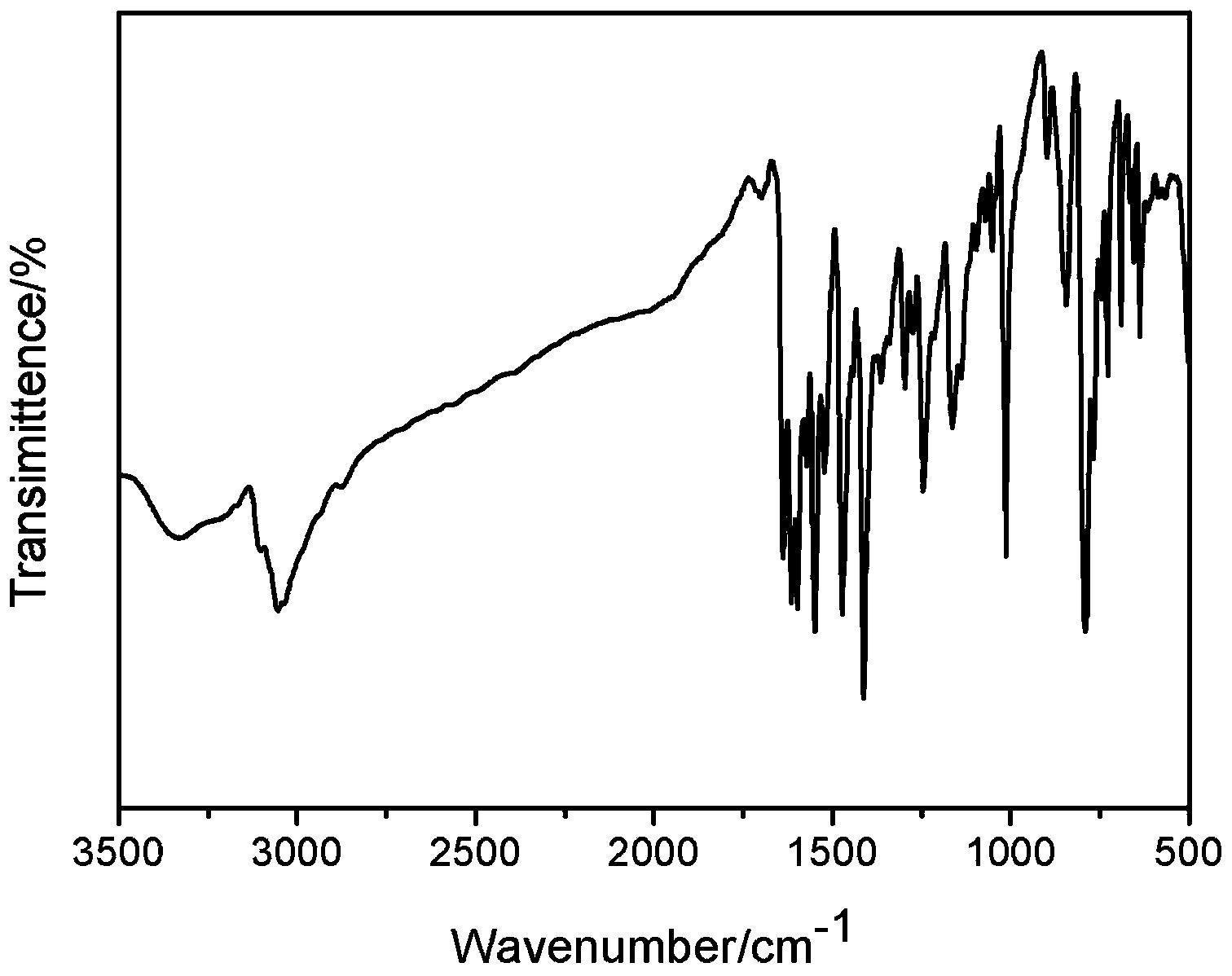
L1La(THF)3数据:产率87%。元素分析:C,58.88;H,6.84;N,1.84;La,18.82。C39H54NO6La理论值:C,60.93;H,6.69;N,1.82;Nd,18.07。红外吸收光谱数据(KBr,cm-1):3425(s),2914(w),1610(s),1478(s),1384(s),1306(s),1257(s),1160(s),1053(s),857(s),802(s),762(s),613(s),603(s)。核磁数据:1H NMR(400MHz,THF-d8):6.73-6.69(s,3H,ArH);6.68-6.65(s,3H,ArH);3.93-3.83(m,3H,THF);2.70-2.59(m,3H,THF);2.16-2.07(d,18H,CH3)。13C NMR(100MHz,THF-d8):162,130,128,123,121,60,20,16。
红外吸收光谱和核磁数据表明目的化合物制备成功。
实施例2:制备L1Nd(THF)3
其步骤(1)、(2)和(3)同实施例1,其中步骤(2)中LaCp3(THF)相应的换成NdCp3(THF),四氢呋喃溶剂换成乙醚。
制备得到的L1Nd(THF)3数据:产率85%。元素分析:C,58.88;H,6.84;N,1.84;Nd,18.90。C39H54NO6Nd理论值:C,60.51;H,6.64;N,1.81;Nd,18.63。红外吸收光谱数据(KBr,cm-1):3411(s),2918(w),1613(s),1478(s),1383(s),1346(s),1310(s),1258(s),1160(s),1056(s),858(s),800(s),759(s),641(s),624(s)。
红外吸收光谱和元素分析数据表明目的化合物制备成功。
实施例3:制备L1Sm(THF)3
其步骤(1)、(2)和(3)同实施例1。步骤(2)中LaCp3(THF)相应换成SmCp3(THF),四氢呋喃溶剂换成乙二醇二甲醚。
本实施例制备的L1Sm(THF)3数据:产率80%。元素分析:C,58.96;H,6.89;N,1.88;Sm,18.85。C39H54NO6Sm理论值:C,60.04;H,6.59;N,1.80;Sm,19.27。红外吸收光谱数据(KBr,cm-1):3390(s),2916(w),1610(s),1478(s),1386(s),1365(s),1307(s),1263(s),1160(s),1054(s),855(s),810(s),760(s),660(s),630(s)。
红外吸收光谱和元素分析表明目的化合物制备成功。
实施例4:制备L1Y(THF)3
其步骤(1)、(2)和(3)同实施例1。步骤(2)中LaCp3(THF)相应的换成YCp3(THF)。
本实施例制备的L1Y(THF)3数据:产率64%。元素分析:C,63.55;H,6.90;N,1.78;Y,12.26。C39H54NO6Y理论值:C,65.17;H,7.15;N,1.95;Y,12.37。核磁数据:1H NMR(400MHz,THF-d8):6.74-6.70(s,3H,ArH);6.70-6.64(s,3H,ArH);3.95-3.80(m,3H,THF);2.75-2.55(m,3H,THF);2.18-2.06(d,18H,CH3)。13C NMR(100MHz,THF-d8):160,130,128,123,121,60,20,16。红外吸收光谱数据(KBr,cm-1):3380(s),2910(w),1606(s),1469(s),1378(s),1340(s),1306(s),1250(s),1151(s),1049(s),850(s),801(s),752(s),640(s),631(s),625(s)。以上数据证明目的化合物制备成功。
实施例5:制备L2Nd(THF)3
其步骤如下:
(1)合成配体L2H3,其制备方法如下:
2,2’,2”-[Nitrilotris(methylene)]tris[4,6-dichlorophenol](L2H3)的合成:按40:3.3:14.2的摩尔比取2,4-二氯苯酚、六次甲基四胺和35%的甲醛溶液,放入圆底烧瓶中反应,在140℃下反应4小时后,降至室温,氯仿终止反应,用去离子水洗3次,之后用无水MgSO4干燥,然后浓缩,得到油状物,柱层析分离。先用乙酸乙酯:石油醚=1:100的洗脱剂淋洗,洗去杂质,再用乙酸乙酯:石油醚=1:20的洗脱剂淋洗,浓缩,析出白色固体,抽干,得到化合物L2H3,产率15%。核磁数据:1H NMR(400MHz,CDCl3):7.27-7.24(d,J=2.4Hz,3H,ArH);7.06-7.03(d,J=2.4Hz,3H,ArH);3.76-3.73(s,6H,CH2)。
(2)合成氮桥连三芳氧基稀土金属化合物L2Nd(THF)3
在一除水除氧、氩气保护的反应瓶中加入NdCp3(THF),加入四氢呋喃溶剂,在另一个除水除氧、氩气保护的反应瓶中称取与NdCp3(THF)等摩尔比的配体L2H3,用四氢呋喃溶剂溶解,将配体溶液缓慢加入到NdCp3(THF)的四氢呋喃悬浊液中,溶液逐渐变澄清,反应12小时,最终得到澄清液。
(3)抽干溶剂,再加入四氢呋喃至恰好溶解,离心,取上层清液至结晶瓶中,室温静置直至析出晶体,即为氮桥连三芳氧基稀土金属化合物L2NdCp3(THF)。
本实施例制备的L2Nd(THF)3数据:产率81%。元素分析:C,42.97;H,3.98;Cl,23.45;N,1.70;Nd,16.19。C33H35Cl6NO6Nd理论值:C,44.06;H,4.03;Cl,23.65;N,1.56;Nd,16.03。红外吸收光谱数据(KBr,cm-1):3430(s),2843(w),1620(s),1469(s),1383(s),1335(s),1290(s),1218(s),1169(s),854(s),756(s),570(s)。
红外吸收光谱和元素分析数据表明目的化合物制备成功。
实施例6:制备L3Nd(THF)3
其步骤如下:
(1)合成配体L3H3,其制备方法如下:
2,2',2″-[Nitrilotris(methylene)]tris[6-tert-butyl-4-methylphenol](L3H3)的合成:按40:3.3:14.2的摩尔比取2-叔丁基对甲苯酚、六次甲基四胺和35%的甲醛溶液,放入圆底烧瓶中反应,在120℃反应48小时后,降至室温,氯仿终止反应,用去离子水洗3次,之后用无水MgSO4干燥,然后浓缩,得到油状物,柱层析分离。先用乙酸乙酯:石油醚=1:50的洗脱剂淋洗,洗去杂质,再用乙酸乙酯:石油醚=1:10的洗脱剂淋洗,浓缩,析出粉红色固体,抽干,得到化合物L3H3,产率10%。核磁数据:1H NMR(400MHz,CDCl3):7.05-7.03(m,3H,ArH);6.81-6.78(m,3H,ArH);3.61-3.59(s,6H,CH2);2.27-2.25(s,9H,CH3);1.41-1.38(s,27H,But)。
(2)合成氮桥连三芳氧基稀土金属化合物L3Nd(THF)3
在一除水除氧、氩气保护的反应瓶中加入NdCp3(THF),加入四氢呋喃溶剂,在另一个除水除氧、氩气保护的反应瓶中称取与NdCp3(THF)等摩尔比的配体L3H3,用四氢呋喃溶剂溶解,将配体溶液缓慢加入到NdCp3(THF)的四氢呋喃悬浊液中,溶液逐渐变澄清,反应18小时,最终得到澄清液。
(3)抽干溶剂,再加入四氢呋喃至恰好溶解,离心,取上层清液至结晶瓶中,室温静置直至析出晶体,即为氮桥连三芳氧基稀土金属化合物L3NdCp3(THF)。
本实施例制备的L3Nd(THF)3数据:产率75%。元素分析:C,62.16;H,7.90;N,1.70;Nd,15.69。C48H72NO6Nd理论值:C,63.82;H,8.03;N,1.55;Nd,15.97。红外吸收光谱数据(KBr,cm-1):3401(s),2906(w),1597(s),1460(s),1368(s),1338(s),1299(s),1247(s),1150(s),1049(s),840(s),791(s),750(s),637(s),620(s)。
红外吸收光谱和元素分析数据表明目的化合物制备成功。
实施例7:0.33mol%的L1Nd(THF)3与0.33mol%的四丁基溴化铵在60℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气体保护下,在反应瓶中加入0.0111克(1.43×10-5摩尔)LNd(THF)3,再加入0.0046克(1.43×10-5摩尔)四丁基溴化铵,加入2毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在60℃的恒温浴中搅拌反应,18小时后终止反应。
取一滴反应液进行核磁表征,计算转化率为33%,核磁数据:1H NMR(CDCl3,400MHz):7.60-7.52(d,J=8.0Hz,2H,ArH),7.44-7.36(t,J=7.6Hz,2H,ArH),7.20-7.13(t,J=7.4Hz,1H,ArH),4.85-4.75(sex,J=6.4Hz,1H,CH),4.17-4.11(t,J=8.4Hz,1H,CH2),3.67-3.62(dd,J=8.3,7.4Hz,1H,CH2),1.58-1.53(d,J=6.2Hz,3H,CH3)。
将反应液旋干,通过柱层析法分离出产物。先用乙酸乙酯:石油醚=1:10的洗脱剂洗出杂质,再用乙酸乙酯:石油醚=1:5的洗脱剂分离出产物,分离产率为82%。
实施例8:0.33mol%的L1Nd(THF)3与0.33mol%的四丁基溴化铵在120℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0111克(1.43×10-5摩尔)LNd(THF)3,再加入0.0046克(1.43×10-5摩尔)四丁基溴化铵,加入2毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在120℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为78%。
实施例9:0.33mol%的L1Nd(THF)3与0.33mol%的四丁基溴化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0111克(1.43×10-5摩尔)LNd(THF)3,再加入0.0046克(1.43×10-5摩尔)四丁基溴化铵,加入2毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。24小时后终止反应,取一滴反应液进行核磁表征,计算转化率为52%。
实施例10:0.33mol%的L1Nd(THF)3与0.33mol%的四丁基溴化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0111克(1.43×10-5摩尔)LNd(THF)3,再加入0.0046克(1.43×10-5摩尔)四丁基溴化铵,加入2毫升溴苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为44%。
实施例11:0.33mol%的L1Nd(THF)3与0.33mol%的四丁基溴化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0111克(1.43×10-5摩尔)LNd(THF)3,再加入0.0046克(1.43×10-5摩尔)四丁基溴化铵,加入2毫升二甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为43%。
实施例12:2.0mol%的L1Nd(THF)3与2.0mol%的四丁基碘化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0222克(2.86×10-5摩尔)LNd(THF)3,再加入0.0106克(2.86×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.1毫升(1.43×10-3摩尔)环氧丙烷和0.16毫升(1.43×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。12小时后终止反应,取一滴反应液进行核磁表征,计算转化率为96%。
实施例13:0.33mol%的L1Nd(THF)3与0.67mol%的四丁基碘化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0111克(1.43×10-5摩尔)LNd(THF)3,再加入0.0092克(2.86×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为91%。
实施例14:0.5mol%的L1Y(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0155克(2.14×10-5摩尔)LY(THF)3,再加入0.0158克(4.28×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为96%。
实施例15:0.5mol%的L1Sm(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0167克(2.14×10-5摩尔)LSm(THF)3,再加入0.0158克(4.28×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为96%。
实施例16:0.5mol%的L1Nd(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0166克(2.14×10-5摩尔)LNd(THF)3,再加入0.0158克(4.28×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为98%。
实施例17:0.5mol%的L1La(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0165克(2.14×10-5摩尔)LLa(THF)3,再加入0.0158克(4.28×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为99%。
实施例18:0.5mol%的L1Nd(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧氯丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0247克(3.18×10-5摩尔)LNd(THF)3,再加入0.0235克(6.36×10-5摩尔)四丁基碘化铵,加入8.6毫升甲苯,再分别加入0.5毫升(6.35×10-3摩尔)环氧氯丙烷和0.69毫升(6.35×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌,进行反应。
18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为100%,核磁数据:1H NMR(CDCl3,400MHz):7.65-7.53(d,J=7.9Hz,2H,ArH),7.48-7.38(t,J=7.5Hz,2H,ArH),7.24-7.14(t,J=7.3Hz,1H,ArH),4.95-4.85(m,1H,CH),4.25-4.17(t,J=8.9Hz,1H,CH2),4.04-3.96(dd,J=8.9,5.8Hz,1H,CH2),3.87-3.73(m,2H,CH2Cl)。
将反应液旋干,通过柱层析法分离出产物。先用乙酸乙酯:石油醚=1:10的洗脱剂洗出杂质,再用乙酸乙酯:石油醚=1:5的洗脱剂分离出产物,分离产率为86%。
实施例19:0.5mol%的L1Nd(THF)3与1.0mol%的四丁基溴化铵在80℃下催化氧化苯乙烯和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0170克(3.18×10-5摩尔)LNd(THF)3,再加入0.0141克(6.36×10-5摩尔)四丁基溴化铵,加入6毫升甲苯,再分别加入0.5毫升(4.39×10-3摩尔)氧化苯乙烯和0.475毫升(4.39×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。
18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为100%,4位取代的恶唑烷酮类化合物与5位取代的恶唑烷酮类化合物比例约为0.47:1,核磁数据:1H NMR(CDCl3,400MHz):3,5-二苯基恶唑烷酮:7.61-7.35(m,9H,ArH),7.21-7.15(tt,J=7.4,1.1Hz,1H,ArH),5.71-5.64(dd,J=8.4,8.2Hz,1H,CH),4.46-4.38(t,J=8.8Hz,1H,CH2),4.03-3.97(dd,J=8.8,7.6Hz,1H,CH2);3,4-二苯基恶唑烷酮:7.45-7.20(m,9H,ArH),7.09-7.04(tt,J=7.4,1.1Hz,1H,ArH),5.42-5.36(dd,J=8.7,6.0Hz,1H,CH),4.81-4.75(t,J=8.7Hz,1H,CH2),4.23-3.17(dd,J=8.6,6.0Hz,1H,CH2)。
将反应液旋干,通过柱层析法分离出产物。先用乙酸乙酯:石油醚=1:10的洗脱剂洗出杂质,再用乙酸乙酯:石油醚=1:5的洗脱剂分离出产物,分离产率:3,5-二苯基恶唑烷酮为62%,3,4-二苯基恶唑烷酮为27%。
实施例20:1.0mol%的L1Nd(THF)3与2.0mol%的四丁基碘化铵在80℃下催化环氧环戊烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0267克(3.44×10-5摩尔)LNd(THF)3,再加入0.0254克(6.88×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(3.40×10-3摩尔)环氧环戊烷和0.37毫升(3.40×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为75%,核磁数据:1HNMR(CDCl3,400MHz):7.62-7.53(d,J=8.9Hz,2H,ArH),7.36-7.42(m,2H,ArH),7.19-7.12(tt,J=7.4,2.1Hz,1H,ArH),5.09-5.03(m,1H,CH),4.81-4.73(m,1H,CH),2.25-2.13(m,1H,CH2),2.03-1.95(m,1H,CH2),1.87-1.67(m,4H,CH2)。13C NMR(CDCl3,100MHz):155.2,137.3,129.2,124.2,119.8,78.6,61.1,34.0,31.7,22.2。
将反应液旋干,通过柱层析法分离出产物。先用乙酸乙酯:石油醚=1:10的洗脱剂洗出杂质,再用乙酸乙酯:石油醚=1:5的洗脱剂分离出产物,分离产率为69%。
实施例21:0.5mol%的L1Nd(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧丙烷和对甲苯基异氰酸酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0166克(2.14×10-5摩尔)LNd(THF)3,再加入0.0158克(4.28×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.28×10-3摩尔)环氧丙烷和0.54毫升(4.28×10-3摩尔)对甲苯基异氰酸酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为100%,核磁数据:1H NMR(CDCl3,400MHz):7.46-7.40(d,J=8.6Hz,2H,ArH),7.22-7.17(d,J=8.2Hz,2H,ArH),4.85-4.73(m,1H,CH),4.15-4.08(t,J=8.5Hz,1H,CH2),3.66-3.59(dd,J=8.6,7.0Hz,1H,CH2),2.37-2.32(s,3H,CH3),1.58-1.52(d,J=6.2Hz,3H,CH3)。13C NMR(CDCl3,100MHz):154.9,135.9,133.6,129.5,118.2,69.5,52.1,20.7。
将反应液旋干,通过柱层析法分离出产物。先用乙酸乙酯:石油醚=1:10的洗脱剂洗出杂质,再用乙酸乙酯:石油醚=1:5的洗脱剂分离出产物,分离产率为89%。
实施例22:1.0mol%的L1Nd(THF)3与2.0mol%的四丁基碘化铵在80℃催化环氧丙烷和对氯苯基异氰酸酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0333克(4.28×10-5摩尔)LNd(THF)3,再加入0.0317克(8.56×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.28×10-3摩尔)环氧丙烷和0.8489克(4.28×10-3摩尔)对氯苯基异氰酸酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为78%,核磁数据:1H NMR(CDCl3,400MHz):7.53-7.48(d,J=9.0Hz,2H,ArH),7.37-7.33(d,J=9.0Hz,2H,ArH),4.87-4.77(m,1H,CH),4.15-4.07(t,J=8.4Hz,1H,CH2),3.65-3.59(dd,J=8.6,7.1Hz,1H,CH2),1.58-1.54(d,J=6.3Hz,3H,CH3)。13CNMR(CDCl3,100MHz):154.7,137.0,129.6,129.0,119.5,69.6,51.8,20.6。
将反应液旋干,通过柱层析法分离出产物。先用乙酸乙酯:石油醚=1:10的洗脱剂洗出杂质,再用乙酸乙酯:石油醚=1:5的洗脱剂分离出产物,分离产率为68%。
实施例23:0.5mol%的L1Nd(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧丙醇和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0175克(2.25×10-5摩尔)LNd(THF)3,再加入0.0166克(4.50×10-5摩尔)四丁基碘化铵,加入6.2毫升甲苯,再分别加入0.3毫升(4.39×10-3摩尔)环氧丙醇和0.5毫升(4.39×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为82%,核磁数据:1H NMR(CDCl3,400MHz):7.53-7.37(m,4H,ArH),7.26-7.20(tt,J=7.3,1.2Hz,1H,ArH),4.60-4.45(m,3H,CH,CH2OH),3.90-3.65(m,2H,CH2),1.87-1.80(s,1H,OH)。
将反应液旋干,通过柱层析法分离出产物。先用乙酸乙酯:石油醚=1:1的洗脱剂洗出杂质,再用乙酸乙酯:石油醚=1:5的洗脱剂分离出产物,分离产率为73%。
实施例24:0.5mol%的L2Nd(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0193克(2.14×10-5摩尔)L2Nd(THF)3,再加入0.0158克(4.28×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为94%。
实施例25:0.5mol%的L3Nd(THF)3与1.0mol%的四丁基碘化铵在80℃下催化环氧丙烷和异氰酸苯酯环加成反应:
无水无氧、氩气保护下,在反应瓶中加入0.0194克(2.14×10-5摩尔)L3Nd(THF)3,再加入0.0158克(4.28×10-5摩尔)四丁基碘化铵,加入6毫升甲苯,再分别加入0.3毫升(4.29×10-3摩尔)环氧丙烷和0.47毫升(4.29×10-3摩尔)异氰酸苯酯,在80℃的恒温浴中搅拌反应。18小时后终止反应,取一滴反应液进行核磁表征,计算转化率为90%。
通过上述实施例可以看出本发明中的一种制备恶唑烷酮类化合物的方法,具体为采用氮桥连三芳氧基稀土金属化合物与季铵盐组成的双组分催化体系催化异氰酸酯与环氧烷反应,制备恶唑酮类化合物。所述氮桥连三芳氧基稀土金属化合物的通式为:LLn(THF)3,式中L代表氮桥连三芳氧基配体;Ln表示稀土金属离子,选自La,Nd,Sm,Y中的一种;季铵盐是四丁基碘化铵,四丁基溴化铵,四丁基氯化铵,四辛基溴化铵,双(三苯基膦)氯化铵,苄基三丁基溴化铵中的一种。该催化体系中的稀土金属催化剂结构明确,易于合成,催化活性高,催化剂用量少。本发明公开的制备方法中原料易得,反应条件温和,底物普适性广,目标产物的收率高,反应操作和后处理过程简单。
当然上述实施例只是为说明本发明的技术构思及特点所作的例举而非穷举,其目的在于让熟悉此项技术的人能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明主要技术方案的精神实质所做的修饰,都应涵盖在本发明的保护范围之内。
一种氮桥连三芳氧基稀土金属化合物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。









动态评分
0.0