









专利摘要
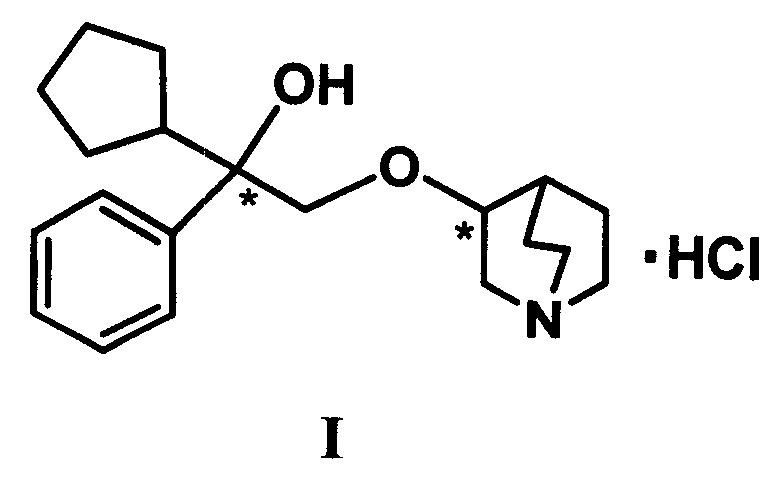
本发明涉及制备式I盐酸戊乙奎醚,即3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷光学异构体的方法,该方法包括以光学纯的(S)或(R)-扁桃酸为手性模板原料,立体选择性合成关键中间体(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷,再与(3S)或(3R)-奎宁环醇进行反应,相应得到目标化合物的四个光学异构体。目标化合物的光学纯度在98%以上。本发明合成方法原料易得,合成过程具有较高的立体选择性,目标产物产率高,且合成成本低。
权利要求
1.制备式I盐酸戊乙奎醚即3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷光学异构体的方法,
该方法包括使光学纯的(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷与(3S)或(3R)-奎宁环醇进行反应。
2.权利要求1所述的方法,其中光学纯的(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷是按照如下方法制备的:
(a)以R或S扁桃酸为手性模板,以甲磺酸、三氟甲磺酸、对甲苯磺酸等为催化剂,在25~45℃下,使扁桃酸与特戊醛反应5~10小时进行醇醛缩合,立体选择性得到内酯产物2-特丁基-5-苯基-1,3-二噁烷-4-酮;
(b)使环戊酮与烯醇形式的2-特丁基-5-苯基-1,3-二噁烷-4-酮在-80~-60℃并于金属锂试剂存在下进行立体控制性的Michael加成反应,得到加成产物2-特丁基-5-苯基-5-(环戊基-1-羟基)-1,3-二噁烷-4-酮;
(c)使步骤(b)的产物经历脱水、脱保护、氢化反应后,得到(S)或(R)-α-苯基-α-环戊基-α-羟基乙酸;后者在醇性溶剂中经Pd/C、Ni-H等催化还原得到(S)或(R)-α-苯基-α-环戊基-α-羟基乙醇,再与甲磺酰氯、苯磺酰氯或对甲苯磺酰氯反应进行单酯化及分子内的闭环反应,得到(S)或(R)-α-苯基-α-环戊基-α-羟基环氧乙烷。
3.权利要求1所述的方法,其中(3S)或(3R)-奎宁环醇先被去质子化,再与(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷反应。
4.权利要求3所述的方法,所述的去质子化是用金属钠或碱金属氢化物进行的。
5.权利要求4所述的方法,其中的碱金属氢化物为氢化钠或氢化钾。
6.权利要求1或2所述的方法,其中制备得到的戊乙奎醚,即3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷光学异构体的纯度在98%以上。
7.光学纯的(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷。
说明书
技术领域技术领域
本发明涉及一种立体选择性合成戊乙奎醚光学异构体的方法。
技术背景背景技术
盐酸戊乙奎醚[Penehyclidine Hydrochloride,即3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐,其结构如下式(I)所示]属于乙基环烃胺基醚类化合物,它是一种新型结构选择性抗胆碱药物(林永煅等.军事医学科学院院刊,1987,11,356.),临床已经成功用于救治有机磷农药中毒等方面,其抗毒效价高,作用持久、不良作用较少(曾繁忠等.中华内科杂志.1993,32,838;乔建忠等.中国药学杂志.2003,38,942.)。在抗胆碱作用的评价研究中,与工具药QNB(3-Quinuclidine benziate)相比具有同等剂量水平的抗神经毒剂作用,其抗槟榔碱所引起的震颤和流诞作用分别比QNB强10倍和2倍(沈淑英等.中国药理学报.1985,6,158.)。现作为药用的为其外消旋体。
盐酸戊乙奎醚的分子结构中含有两个手性中心,所以具有四个光学异构体。初步研究表明,盐酸戊乙奎醚抗胆碱作用的活性异构体为(R,R)-构型。就对大鼠大脑M受体亲和力而言,最强的(R,R)-异构体的作用比最弱的(S,S)-异构体高1000多倍(Niu,W.Z.et al Arch.Int.Pharmacodyn Ther.1990,304,64.)。
通常,戊乙奎醚光学异构体的制备是采用苯甲睛与格氏试剂反应得到苯基环戊基甲酮,然后进一步合成消旋的1,1-苯基环戊基环氧乙烷;再将其分别与3S及3R-奎宁环醇反应,得到两个对非对映异构体;最后经制备薄层色谱技术分离纯化而获得各个光学异构体(高建华等.药学学报.1990,25,891.)。由于分离纯化方法的局限,导致该方法耗时费力,不利于大规模合成。到目前为止,尚无有关从光学纯原料立体定向选择性合成戊乙奎醚光学异构体方法的报道,因此迫切需要开发戊乙奎醚光学异构体的立体定向选择性合成方法。
发明内容发明内容
本发明的目的是提供一种原料易得、反应条件温和、立体选择性高的合成3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷四个光学异构体(3S,2′S),(3S,2′R),(3R,2′R)及(3R,2′S)(下文中分别以式Ia、Ib、Ic、Id表示)的方法。
因此,本发明的一个方面涉及立体选择性合成3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷四个光学异构体的方法,该方法包括使光学纯的(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷与(3S)或(3R)-奎宁环醇进行反应。
本发明的另一方面涉及作为戊乙奎醚光学异构体立体选择性合成的关键中间体光学纯的(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷的制备方法。
本发明进一步的方面涉及作为戊乙奎醚立体选择性合成关键中间体的光学纯的(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷。
本发明的上述目的可以通过下列合成路线来实现:以S或R扁桃酸为手性模板,使扁桃酸分子中的羟基先与特戊醛进行醇醛缩和,形成的半缩醛分子中的羟基再与羧基进行分子内的缩合,立体选择性得到2-特丁基-5-苯基-1,3-二噁烷-4-酮(IIa或IIb);然后,使环戊酮与烯醇形式的IIa或IIb进行立体控制性的Michael加成反应,得到的加成产物2-特丁基-5-苯基-5-(环戊基-1-羟基)-1,3-二噁烷-4-酮(IIIa或IIIb),经脱水、脱保护、氢化,即可得到(S)或(R)-α-苯基-α-环戊基-α-羟基乙酸(VIa或VIb);后者经还原产物二醇的单磺酰化以形成S或R-α-苯基-α-环戊基-α羟基对甲苯磺酸单酯(VIIIa或VIIIb),再经过分子内环化反应得到光学纯度在99%以上的合成关键中间体(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷(IXa或IXb),其再与(3S)或(3R)-奎宁环醇进行反应,即得到目标化合物。任选地,经盐酸化得到目标化合物的盐酸化产物。
下面以式Ia化合物的制备为例,给出具体的合成路线图示:
因此,根据本发明,合成3-(2-羟基-2-环戊基-2-苯基乙氧基)奎宁环烷四个光学异构体(Ia、Ib、Ic、Id)所用到的关键性中间体光学纯的(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷的制备依次包括以下步骤:
(a)以R或S扁桃酸为手性模板,以甲磺酸、三氟甲磺酸、对甲苯磺酸等为催化剂,在25~45℃下,使扁桃酸分子中的羟基先与特戊醛进行醇醛缩合,形成的半缩醛分子中的羟基再与羧基进行分子内的缩合,反应5~10小时,立体选择性得到内酯产物2-特丁基-5-苯基-1,3-二噁烷-4-酮(IIa或IIb),反应溶剂可以用正戊烷、环己烷、正己烷、苯等非极性溶剂。
(b)使环戊酮与烯醇形式的2-特丁基-5-苯基-1,3-二噁烷-4-酮(IIa或IIb)在-80~-60℃并于金属锂试剂例如LDA、二(三甲硅基)氨基锂等存在下,进行立体控制性的Michael加成反应,得到加成产物2-特丁基-5-苯基-5-(环戊基-1-羟基)-1,3-二噁烷-4-酮(IIIa或IIIb)。
(c)使步骤(b)得到的产物经历脱水、脱保护、氢化反应后,得到(S)或(R)-α-苯基-α-环戊基-α-羟基乙酸(VIa或VIb);后者可以在醇性溶剂例如甲醇、乙醇中经Pd/C、Ni-H等催化还原得到(S)或(R)-α-苯基-α-环戊基-α-羟基乙醇(VIIa或VIIb),其再与甲磺酰氯、苯磺酰氯、对甲苯磺酰氯等反应进行单酯化及分子内的闭环反应,得到光学纯度在98%以上的合成关键中间体,(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷(IXa或IXb)。
反应中所用到的原料(3S)或(3R)-奎宁环醇可以通过消旋体3-奎宁环醇的乙酸酯经光学纯酒石酸拆分得到(B.Ringdahl,B.Resul,R.Dahlbom,Acta.Pharm.Suec.1979,16,281.)。
(3S)或(3R)-奎宁环醇可以任选先经金属钠或碱金属氢化物如氢化钠、氢化钾等去质子化,再在二甲基亚砜溶剂中与(S)或(R)-α-苯基-α-环戊基-1,2-环氧乙烷反应,制备得到前述式Ia-Id戊乙奎醚光学异构体。由此制得的目标产物戊乙奎醚光学异构体的纯度也在98%以上。
附图说明具体实施方式具体实施方式
下面用实施例具体说明本发明,这些实施例不应理解为在任何意义上对本发明构成限制。
实施例1、(2S,5S)-2-特丁基-5-苯基-1,3-二噁烷-4-酮(IIa)的制备
将S-扁桃酸20g(0.13mol)加入200mL正戊烷中,加入21.2mL特戊醛(含量80%,0.16mol,购自Fluka公司),然后加入0.5mL三氟甲磺酸,回流6小时,以分水器除去生成的水。冷却至室温,加入100mL 8%碳酸氢钠溶液,减压蒸除正戊烷,过滤,得到(2S,5S)-2-特丁基-5-苯基-1,3-二噁烷-4-酮(IIa),产量27.1g,熔点100-102℃,产率95%,元素分析,理论值%:C 70.89,H 7.32;实验值%:C 70.81,H 7.39。1H-NMR:δ(ppm,CDCl3),1.10(s,9H),5.25(s,1H),5.38(s,1H),7.47(m,5H)。
实施例2、(2R,5R)-2-特丁基-5-苯基-1,3-二噁烷-4-酮(IIb)的制备
参照实施例1的方法,以(R)-扁桃酸为原料,合成(2R,5R)-2-特丁基-5-苯基-1,3-二噁烷-4-酮(IIb),熔点100-102℃,产率97%,元素分析,理论值%:C 70.89,H 7.32;实验值%:C 70.83 H 7.30。1H-NMR:δ(ppm,CDCl3),1.12(s,9H),5.23(s,1H),5.36(s,1H),7.49(m,5H)。
实施例3、(2S,5R)-2-特丁基-5-苯基-5-(环戊基-1-羟基)-1,3-二噁烷-4-酮(IIIa)的制备
将10g(45mmol)(2S,5S)-2-特丁基-5-苯基-1,3-二噁烷-4-酮溶于70mL干燥四氢呋喃中,冷却至-78℃,加入60mL二(三甲硅基)氨基锂(四氢呋喃溶液,1.0M)。搅拌下滴加65mmol环戊酮,搅拌反应2小时,滴加15mL饱和磷酸氢钠溶液和200mL饱和氯化铵溶液。分出有机层,水层用乙酸乙酯萃取,合并有机层,干燥,蒸除溶剂,得到(2S,5R)-2-特丁基-5-苯基-5-(环戊基-1-羟基)-1,3-二噁烷-4-酮(IIIa),产量10.5g,产率74%。1H-NMR:δ(ppm,CDCl3),0.88(s,9H),1.52-2.06(m,8H),5.52(s,1H),7.31(m,3H),7.78(dd,J=1.5,8.3Hz,2H)。
实施例4、(2R,5S)-2-特丁基-5-苯基-5-(环戊基-1-羟基)-1,3-二噁烷-4-酮(IIIb)的制备
参照实施例3的方法,使(2R,5R)-2-特丁基-5-苯基-1,3-二噁烷-4-酮(IIb)与环戊酮反应,得到(2R,5S)-2-特丁基-5-苯基-5-(环戊基-1-羟基)-1,3-二噁烷-4-酮(IIIb),产率71%。1H-NMR:δ(ppm,CDCl3),0.89(s,9H),1.51-2.07(m,8H),5.54(s,1H),7.30(m,3H),7.76(dd,J=1.5,8.3Hz,2H)。
实施例5、(2S,5S)-2-特丁基-5-苯基-5-(1-环戊烯基)-1,3-二噁烷-4-酮(IVa)的制备
将3.6g(10mmol)(2S,5R)-2-特丁基-5-苯基-5-(环戊基-1-羟基)-1,3-二噁烷-4-酮溶于70mL干燥四氢呋喃溶液中,冷却至0℃,加入2mL氯化亚砜和3mL吡啶,搅拌反应1小时。加入60mL饱和氯化铵溶液,分出有机层,水层用乙酸乙酯萃取,合并有机层,干燥,蒸除溶剂,得到(2S,5S)-2-特丁基-5-苯基-5-(1-环戊烯基)-1,3-二噁烷-4-酮(IVa),产量3.7g,产率95%。1H-NMR:δ(ppm,CDCl3),1.07(s,9H),1.92-2.50(m,6H),5.22(s,1H),6.03(m,1H),7.25(m,4H),7.59(m,1H)。
实施例6、(2R,5R)-2-特丁基-5-苯基-5-(1-环戊烯基)-1,3-二噁烷-4-酮(IVb)的制备
参照实施例5的方法,由IIIb得到(2R,5R)-2-特丁基-5-苯基-5-(1-环戊烯基)-1,3-二噁烷-4-酮(IVb),产率92%。1H-NMR:δ(ppm,CDCl3),1.05(s,9H),1.90-2.51(m,6H),5.23(s,1H),6.01(m,1H),7.28(m,4H),7.57(m,1H)。
实施例7、(S)-α-苯基-α-环戊基-α-羟基乙酸(VIa)的制备
将2.34g(8.18mmol)(IVa)溶于20mL甲醇和10mL水混合溶液,加入4.58g KOH,搅拌回流反应3小时。冷却至室温,正庚烷萃取,水相用1NHCl酸化,乙酸乙酯萃取,干燥,蒸除溶剂得(S)-α-苯基-α-环戊烯基-α-羟基乙酸(Va),产量1.64g,产率92%,1H NMR(CDCl3)1.81(m,2H),2.29(m,4H),5.59(s,1H),7.24(t,J=6.8Hz,1H),7.32(t,J=7.1Hz,2H),7.48(d,J=7.8Hz,1H)。将1.16g(5.3mmol)(S)-α-苯基-α-环戊烯基-α-羟基乙酸(Va)溶于50mL甲醇中,加入0.2g10%Pd/C,1大气压下用氢气还原8个小时。过滤,减压蒸除溶剂,得到1.08g(S)-α-苯基-α-环戊基-α-羟基乙酸(VIa),收率93%。[α]D20=-2.5°(MeOH,c=3),熔点:118-119℃。
实施例8、(R)-α-苯基-α-环戊基-α-羟基乙酸(VIb)的制备
参照实施例7的方法,合成(R)-α-苯基-α-环戊基-α-羟基乙酸(VIb),产率93%,[α]D20=+2。4°(MeOH,c=3),熔点:118-120℃。
实施例9、(S)-α-苯基-α-环戊基-α-羟基对甲苯磺酸乙酯(VIIIa)的合成
N2保护下,将含有2.2g(0.01mol)(S)-α-苯基-α-环戊基-α-羟基乙酸(VIa)的10mL无水THF溶液缓慢滴加到含LiAlH4 0.02mol的20mL无水THF溶液中,搅拌下缓慢升温至回流,反应3h。冷却,小心滴加2mL饱和NaHCO3溶液,10mL 2N的NaOH溶液,分出有机相,水相用乙醚提取,合并有机相,饱和食0盐水洗涤,无水硫酸钠干燥。减压蒸除溶剂,得到(S)-α-苯基-α-环戊基-α-羟基乙醇(VIIa)无色针状晶体,2.06g,收率100%,熔点:49-50℃。1H NMR(CDCl3)1.23-1.71(m,8H),2.02(brs,1H),2.20-2.28(m,1H),3.76(d,J=11Hz,1H),3.94(d,J=11Hz,1H),7.23-7.27(m,1H),7.34-7.44(m,4H)。
在干燥的100mL三口瓶中,加入1.89g(S)-α-苯基-α-环戊基-α-羟基乙醇(VIIa)和50mL干燥的CH2Cl2,冷却至0℃,加入3.42g(18mmol)对甲苯磺酸。搅拌下滴加三乙胺2.43g(24mmol)。滴加完毕,于0℃搅拌2h,反应混和液略呈黄色,小心加入饱和NaHCO3,旋转蒸发除去CH2Cl2,乙醚萃取,干燥,石油醚重结晶,得到(S)-α-苯基-α-环戊基-α-羟基对甲苯磺酸乙酯(VIIIa),白色絮状固体,2.61g,收率79%。1HNMR(CDCl3)1.21-1.27(m,2H),1.35-1.70(m,6H),2.17(s,1H),2.24(m,1H)2.40(s,3H),7.21-7.27(m,8H),7.58(d,J=8Hz 2H)。
实施例10、(R)-α-苯基-α-环戊基-α-羟基对甲苯磺酸乙酯(VIIIb)的合成
参照实施例9的方法,合成(R)-α-苯基-α-环戊基-α-羟基对甲苯磺酸乙酯(VIII b),收率78%。1H NMR(CDCl3)1.21-1.27(m,2H),1.35-1.70(m,6H),2.17(s,1H),2.24(m,1H)2.40(s,3H),7.21-7.27(m,8H),7.58(d,J=8Hz 2H)。
实施例11(S)-α-苯基-α-环戊基-1,2-环氧乙烷(IXa)的制备
在干燥的50mL三口瓶中,加入1.0g(S)-α-苯基-α-环戊基-α-羟基对甲苯磺酸乙酯(VIIIa)和20mL无水甲醇,搅拌下,加入过量无水K2CO3。室温搅拌反应30分钟,加水稀释,乙醚萃取,干燥,蒸除溶剂,得到((S)-α-苯基-α-环戊基-1,2-环氧乙烷(IXa),其为无色液体,产量0.5g,收率100%。1H NMR(CDCl3)1.25-1.68(m,8H),2.58-2.63(m,1H),2.66(d,J=5Hz,1H),2.97(d,J=5Hz,1H),7.23-7.40(m,5H)。
实施例12(R)-α-苯基-α-环戊基-1,2-环氧乙烷(IXb)的合成
参照实施例11的方法,合成(R)-α-苯基-α-环戊基-1,2-环氧乙烷(IXb)。收率100%,1H NMR(CDCl3)1.25-1.68(m,8H),2.58-2.63(m,1H),2.66(d,J=5Hz,1H),2.97(d,J=5Hz,1H),7.23-7.40(m,5H)。
实施例13(3S,2′S)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐(Ia)的合成
氮气保护下,将0.6g(15mmol)NaH置于干燥的三口烧瓶中,加入10mL无水DMSO,搅拌5分钟后,滴加含有1.8g(14.2mmol)3S-奎宁环醇的10mLDMSO溶液,混合物于60℃搅拌反应1h。冷至室温,缓慢滴加2.61g(14mmol)(S)-α-苯基-α-环戊基-1,2-环氧乙烷(IXa)的10mL DMSO溶液。在50℃搅拌反应混合物3h,冷却,小心滴加20mL水。乙醚提取,水洗,10%盐酸溶液洗涤醚层,合并水相;40%NaOH碱化盐酸提取液,乙醚萃取,无水硫酸钠干燥,蒸除溶剂后,得到(3S,2′S)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷游离碱,无色液体。将游离碱溶于干燥的无水乙醚中,然后滴加盐酸/乙醚溶液,立即生成白色沉淀,丙酮重结晶,得到(3S,2′S)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐(Ia)。产量3.02g,收率69%。[α]D20=-8.18°(MeOH,c=0.40)。1H NMR(CDCl3)1.23-1.75(m,13H),1.96-1.99(m,2H),2.26-2.32(m,1H),2.52-2.77(m,5H),2.71(s,-OH,1H),2.89-2.95(m,1H),3.35-3.38(m,1H),3.58(d,J=9Hz,1H),3.71(d,J=9Hz,1H),7.29-7.42(m,5H)。
实施例14(3S,2′R)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐(Ib)的合成
参照实施例13的方法,由3S-奎宁环醇和(R)-α-苯基-α-环戊基-1,2-环氧乙烷(IXb)进行合成,得到(3S,2′R)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷游离碱,将游离碱溶于干燥的无水乙醚中,然后滴加盐酸/乙醚溶液,立即生成白色沉淀,丙酮重结晶,得到(3S,2′R)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐(Ib)。收率65%。[α]D20=+44.3°(MeOH,c=0.60)。1H NMR(CDCl3)1.20-1.73(m,13H),1.98-1.99(m,1H),2.26-2.32(m,1H),2.52-2.77(m,5H),2.83(s,-OH,1H),2.93-2.96(m,1H),3.38-3.40(m,1H),3.59(d,J=9Hz,1H),3.71(d,J=9Hz,1H),7.20-7.44(m,5H)。
实施例15(3R,2′R)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐(Ic)的合成
参照实施例13的方法,3R-奎宁环醇和(R)-α-苯基-α-环戊基-1,2-环氧乙烷(IXb)进行合成,得到(3R,2′R)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷游离碱,将游离碱溶于干燥的无水乙醚中,然后滴加盐酸/乙醚溶液,立即生成白色沉淀,丙酮重结晶,得到(3R,2′R)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐(Ic)。收率65%。[α]20D=+8.26°(MeOH,c=0.35)。1H NMR(CDCl3)1.23-1.75(m,13H),1.96-1.99(m,2H),2.26-2.32(m,1H),2.52-2.77(m,5H),2.71(s,-OH,1H),2.89-2.95(m,1H),3.35-3.38(m,1H),3.58(d,J=9Hz,1H),3.71(d,J=9Hz,1H),7.29-7.42(m,5H)。
实施例16(3R,2′S)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐(Id)的合成
参照实施例13的方法,3R-奎宁环醇和(S)-α-苯基-α-环戊基-1,2-环氧乙烷(IXb)进行合成,得到(3R,2′S)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷游离碱,将游离碱溶于干燥的无水乙醚中,然后滴加盐酸/乙醚溶液,立即生成白色沉淀,丙酮重结晶,得到(3R,2′S)-3-(2′-羟基-2′-环戊基-2′-苯基乙氧基)奎宁环烷盐酸盐(Id)。收率68%。[α]20D=-48.1°(MeOH,c=0.50)。1H NMR(CDCl3)1.20-1.73(m,13H),1.98-1.99(m,1H),2.26-2.32(m,1H),2.52-2.77(m,5H),2.83(s,-OH,1H),2.93-2.96(m,1H),3.38-3.40(m,1H),3.59(d,J=9Hz,1H),3.71(d,J=9Hz,1H),7.20-7.44(m,5H)。
戊乙奎醚光学异构体的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




动态评分
0.0