






专利摘要
本发明提供了一种齐拉西酮的制备方法,所述方法是由结构式如(I)的化合物:5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮,在有机酸溶剂中用还原剂进行还原制得所述齐拉西酮,所述的溶剂为含羧基官能团并至少含一个卤原子取代的C1~C6的烷烃酸;本发明所述的齐拉西酮制备方法的有益效果主要体现在:收率高,可达85%以上,操作简单,反应时间短,大为降低了成本,且只需要调节pH值就可以得到纯度很高的产物,利于工业化生产。
权利要求
1.一种齐拉西酮的制备方法,其特征在于所述方法是由结构式如(I)的化合物:5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮,在有机酸溶剂中用还原剂进行还原制得所述齐拉西酮,所述的有机酸溶剂为含羧基官能团并至少含一个卤原子取代的C1~C6的烷烃酸;
2.如权利要求1所述的齐拉西酮的制备方法,其特征在于所述还原剂为三甲基硅烷或三乙基硅烷。
3.如权利要求1所述的齐拉西酮的制备方法,其特征在于所述的有机酸溶剂为下列之一:三氯乙酸、或三氟乙酸、或三氯丙酸。
4.如权利要求1所述的齐拉西酮的制备方法,其特征在于所述还原反应在20~50℃下反应12~28小时,所述5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮与三烷基硅烷物质的量之比为1∶1~6。
5.如权利要求1~4之一所述的齐拉西酮的制备方法,其特征在于所述5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮由如下方法得到:
将结构式为式(II)的化合物:N-(1,2-苯并异噻唑-3-基)哌嗪或其氢卤酸盐,和通式(III)的化合物
在碱的存在下,以碱金属碘化物做催化剂,在含亚砜或酰胺基团的有机溶剂存在下进行缩合反应得到所述的5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮;其中,式(II)L为离去基团,所述离去基团为下列之一:①氯、②溴、③碘、④甲磺酰氧基、⑤对甲苯磺酰氧基。
6.如权利要求5所述的齐拉西酮的制备方法,其特征在于所述的有机溶剂为:二甲基甲酰胺、或二甲基亚砜、或二甲基乙酰胺、或二甲基甲酰胺、或二甲基亚砜、或二甲基乙酰胺各自与下列之一的溶剂的任意比例的混合:醇类、酮类、醚类、或水。
7.如权利要求5所述的齐拉西酮的制备方法,其特征在于所述的碱为有机碱或者无机碱,所述的有机碱为三乙胺或二乙胺;所述的无机碱为碱金属的碳酸盐、碱金属的碳酸氢盐、或碱金属的氢氧化物。
8.如权利要求7所述的齐拉西酮的制备方法,其特征在于所述的碱为碳酸钠、碳酸钾、碳酸氢钠或碳酸氢钾,所述的碱金属碘化物为碘化钠或碘化钾。
9.如权利要求5所述的齐拉西酮的制备方法,其特征在于所述化合物(II)或其盐与化合物(III)物质的量之比为1~2∶1,所述缩合反应中反应温度为25~150℃,反应时间为1~24小时。
10.如权利要求5所述的齐拉西酮的制备方法,其特征在于所述方法如下:
(1)将结构式为式(II)的化合物N-(1,2-苯并异噻唑-3-基)哌嗪或其氢卤酸盐,和通式(III)的化合物
在碱碳酸钠、碳酸钾、碳酸氢钠或碳酸氢钾的存在下,以碘化钠或碘化钾作催化剂,在有机溶剂二甲基甲酰胺、或二甲基亚砜、或二甲基乙酰胺、或者二甲基甲酰胺、或二甲基亚砜、或二甲基乙酰胺各自与水混溶的溶液的存在下,于70~100℃条件下进行2~7小时缩合反应,经分离纯化得到所述的5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮;其中,式(II)中L为氯;
(2)将5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮,在有机酸溶剂三氯乙酸、或三氟乙酸、或三氯丙酸中,与三甲基硅烷或三乙基硅烷在20~50℃下反应12~28小时,制得所述齐拉西酮,所述5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮与三烷基硅烷物质的量之比为1∶1~6。
说明书
技术领域(一)技术领域
本发明涉及一种齐拉西酮的制备方法。
技术背景(二)背景技术
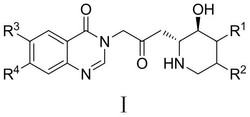
齐拉西酮(ziprasidone)是一种较新的非典型抗精神病药,它是5-羟色胺和多巴胺D2受体的拮抗剂,它能有效治疗精神分裂症阴性和阳性症状,包括幻觉、错觉和主动性缺乏等,由美国Pfizer公司研制,2000年9月首次在瑞典上市,齐拉西酮的化学名称为:5-(2-(4-(1,2-苯并异噻唑-3-基)-1-哌嗪基)乙基)-6-氯-1,3-二氢-2H-吲哚-2-酮,结构式如下:
美国专利US4831031公开了制备齐拉西酮的方法:
该方法是以碘化钠为催化剂,在碳酸钠存在下,甲基异丁基甲酮为溶剂,化合物N-(1,2-苯并异噻唑-3-基)哌嗪和化合物5-(2-氯乙基)6-氯-吲哚酮回流反应40小时,后经柱层析制得齐拉西酮。总收率为20%,这么低的收率说明有大量的副产物,需要昂贵的纯化工序,另外,有许多的报道提到改换溶剂来优化该反应,但是都没有很满意的结果。
发明内容(三)发明内容
为解决现有技术中齐拉西酮制备收率低、操作复杂、反应时间长、成本高的不足,本发明提供了一种收率高、操作简单、反应时间短、成本低的齐拉西酮的制备方法。
为达到发明目的本发明采用的技术方案是:
一种齐拉西酮的制备方法,所述方法是由结构式如(I)的化合物:5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮,在有机酸溶剂中用还原剂进行还原制得所述齐拉西酮,所述的有机酸溶剂为含羧基官能团并至少含一个卤原子取代的C1~C6的烷烃酸;
用该还原方法得到的齐拉西酮,后处理非常简单,纯度很高。
所述还原剂可为三烷基硅烷,所述的三烷基硅烷的烷基为C1~C6的烷基,优选为三甲基硅烷或三乙基硅烷。所述的有机酸溶剂优选为下列之一:三氯乙酸、或三氟乙酸、或三氯丙酸。
所述还原反应在20~50℃下反应12~28小时,所述5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮与三烷基硅烷物质的量之比为1∶1~6。
所述5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮由如下方法得到:
将结构式为式(II)的化合物:N-(1,2-苯并异噻唑-3-基)哌嗪或其氢卤酸盐,和通式(III)的化合物
在碱的存在下,以碱金属碘化物做催化剂,在含亚砜或酰胺基团的有机溶剂存在下进行缩合反应得到所述的5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮;其中,式(II)L为离去基团,所述离去基团为下列之一:①氯、②溴、③碘、④甲磺酰氧基、⑤对甲苯磺酰氧基。
所述的有机溶剂为:二甲基甲酰胺、或二甲基亚砜、或二甲基乙酰胺、或二甲基甲酰胺、或二甲基亚砜、或二甲基乙酰胺各自与下列之一的溶剂的任意比例的混合:醇类、酮类、醚类、或水。
所述的碱为有机碱或者无机碱,所述的有机碱为三乙胺或二乙胺;所述的无机碱为碱金属的碳酸盐、碱金属的碳酸氢盐、或碱金属的氢氧化物。
所述的碱优选为碳酸钠、碳酸钾、碳酸氢钠或碳酸氢钾。所述的碱金属碘化物优选为碘化钠或碘化钾。
所述化合物(II)或其盐与化合物(III)物质的量之比为1~2∶1,所述缩合反应中反应温度为25~150℃,反应时间为1~24小时。
具体的,所述方法如下:
(1)将结构式为式(II)的化合物N-(1,2-苯并异噻唑-3-基)哌嗪或其氢卤酸盐,和通式(III)的化合物
在碱碳酸钠、碳酸钾、碳酸氢钠或碳酸氢钾的存在下,以碘化钠或碘化钾作催化剂,在有机溶剂二甲基甲酰胺、或二甲基亚砜、或二甲基乙酰胺、或者二甲基甲酰胺、或二甲基亚砜、或二甲基乙酰胺各自与水混溶的溶液的存在下,于70~100℃条件下进行2~7小时缩合反应,经分离纯化得到所述的5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮;其中,式(II)中L为氯;
(2)将5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮,在有机酸溶剂三氯乙酸、或三氟乙酸、或三氯丙酸中,与三甲基硅烷或三乙基硅烷在20~50℃下反应12~28小时,制得所述齐拉西酮,所述5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮与三烷基硅烷物质的量之比为1∶1~6。
化合物(II)的制备方法参见欧洲专利EP741129,化合物(III)的制备方法参见期刊J.Med.Chem.1996,39,143-148。
本发明所述的齐拉西酮的新的制备方法及其中间体的有益效果主要体现在:收率高,可达85%以上,操作简单,反应时间短,大为降低了成本,且只需要调节pH值就可以得到纯度很高的产物,利于工业化生产。
附图说明具体实施方式(四)具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
实施例1(对比例):
0.73g(3.20mmol)5-(2-氯乙基)6-氯-吲哚酮,0.70g(3.2mmol)N-(1,2-苯并异噻唑-3-基)哌嗪,0.68g(.40mmol)碳酸钠,2mg碘化钠,30ml甲基异丁基甲酮,加入到装有氮气进口和冷凝器的125ml圆底烧瓶中,反应回流40hr,冷却,过滤,蒸馏。蒸馏物经柱层析纯化,用11乙酸乙酯洗脱副产品,用含4%甲醇的乙酸乙酯1.51洗脱产品,将洗下来的产品蒸馏,将蒸馏物加入到二氯甲烷中,再加入乙醚,通盐酸气使之饱和,产生沉淀,过滤,用乙醚洗涤,过滤,滤饼再用丙酮洗涤,过滤,得到齐拉西酮盐酸盐,收率为20%。
实施例2:5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮的制备:
将N-(1,2-苯并异噻唑-3-基)哌嗪盐酸盐7.9g(31mmol)加入250ml三口烧瓶中,加入40mlDMF(二甲基甲酰胺),搅拌。加入5-(2-氯乙酰基)6-氯-吲哚酮4.88g(20mmol),碘化钾6g,碳酸钾10g,开始升温到70℃,保温4小时后,减压蒸馏,加入40ml水,加入6N盐酸,调PH=3~4,过滤,水洗滤饼至中性,55℃干燥得7.21g产物,收率:82.5%。5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮的物性参数:
MS(%) M+1 426+1
NMR(d,CDCl3):3.2-3.6(m,8H),7.4-7.7(m,2H),7.8(d,1H),7.87(d,1H),2.7(s,2H),6.9(s,1H),10.8(s,1H),3.8(s,2H),7.7(s,1H)
实施例3:5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮的制备:
将N-(1,2-苯并异噻唑-3-基)哌嗪6.8g(31mmol)加入250ml三口烧瓶中,加入30mlDMF(二甲基甲酰胺),搅拌。加入5-(2-溴乙酰基)6-氯-吲哚酮4.88g(20mmol),碘化钠7g,碳酸氢钠11g,开始升温到50℃,保温12小时后,减压蒸馏,加入40ml水,加入6N盐酸,调PH=3~4,过滤,水洗滤饼至中性,55℃干燥得6.02g产物,收率:68.9%。实施例4:5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮的制备:
将N-(1,2-苯并异噻唑-3-基)哌嗪盐酸盐17.3g(67.6mmol)加入250ml三口烧瓶中,加入60mlDMSO(二甲基亚砜),搅拌。加入5-(2-甲磺酰氧基-乙酰基)6-氯-吲哚酮15.1g(50mmol),碘化钾7g,氢氧化钾22g,开始升温到60℃,保温8小时后,加入200ml冰水,析出固体,静置,过滤,水洗滤饼至中性,得红色固体,干燥得13.4g产物,收率:69.5%。
实施例5:5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮的制备:
将N-(1,2-苯并异噻唑-3-基)哌嗪盐酸盐20g(78.3mmol)加入250ml三口烧瓶中,加入60mlDMAC(二甲基乙酰胺),搅拌。加入5-(2-碘乙酰基)6-氯-吲哚酮11g(45mmol),碘化钠8g,碳酸钠26g,开始升温到120℃,保温2小时后,加入200ml冰水,析出固体,静置,过滤,水洗滤饼至中性得红色固体,干燥得15g产物,收率:77.6%。
实施例6:齐拉西酮的制备:
将7.21g(8.6mmol)5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮投入250ml三口瓶,加入60ml三氟乙酸,3.5ml(21.5mmol)三乙基硅烷,升温至30℃反应23小时,减压蒸馏,冷却至0℃,加入饱和碳酸氢钠溶液调PH=7,冷至0℃过滤,得暗红色固体,50℃左右干燥,得6.4g深红色固体,收率:92.1%。5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮与三乙基硅烷的当量比为1∶2.5。
实施例7:齐拉西酮的制备:
取17g(20.3mmol)5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮投入250ml三口瓶,加入80ml三氯乙酸,2.03ml(27.4mmol)三甲基硅烷,升温至50℃反应16小时,减压蒸馏,冷却至0℃,加入饱和碳酸氢钠溶液调PH=7,冷至0℃过滤,得暗红色固体,50℃左右干燥,得13.9g深红色固体,收率:84.5%。5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮与三甲基硅烷的当量比为1∶1.35。
齐拉西酮的物性数据如下:mp>300℃.1H-NMR(δ,DMSO-d6):3.15-3.60(m,10H),3.72(d,2H),4.10(d,2H),6.88(s,1H),7.28(s,1H),7.48(t,1H),7.60(t,1H),8.14(dd,2H),10.6(s,1H),11.4(bs,1H).MS(m/e,%):412(M+,0.4),233(18),232(100),177(19). IR(KBr,cm-1)1708,1628,1489。
实施8:齐拉西酮的制备
取17g(20.3mmol)5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮投入250ml三口瓶,加入100ml三氯丙酸,3.3ml(20.3mmol)三乙基硅烷,升温至50℃反应20小时,减压蒸馏,冷却至0℃,加入饱和碳酸氢钠溶液调PH=7,冷至0℃过滤,得暗红色固体,50℃左右干燥,得13.1g深红色固体,收率:79.5%。5-(2-(4-(1,2-苯并异噻唑-3-基)-哌嗪)乙酰基)-6-氯-1,3-二氢-2H-吲哚-2-酮与三乙基硅烷的当量比为1∶1。
一种齐拉西酮的制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。






![3,7-二[2-(5-溴噻吩基)]-1,5,2,4,6,8-二硫四氮唑辛及其制法和应用](https://www.zhichawang.com/images/CN104356128A/CN104356128A.jpg)










动态评分
0.0