








专利摘要
本发明属于医药技术领域,涉及新的糖肽类抗菌素,具体涉及式1的去甲万古霉素类二聚体衍生物、其药学上可接受的盐、及其制备方法,和所述化合物在用于制备治疗或预防细菌感染性疾病的药物中的用途,本发明中进行了体外抑菌实验,结果表明,所述化合物对多种格兰氏阳性菌的抑菌活性显著高于万古霉素和去甲万古霉素。本发明的化合物可进一步用于制备药物,尤其是制备抗细菌感染的药物。桥链X可以为:其中,R代表氢或取代或非取代的C1‑12的烷基、环烷基、杂环烷基、芳香基、杂芳香取代基,以及C1‑12的烷基酰基、芳香基酰基和烷基磺酰基。
权利要求
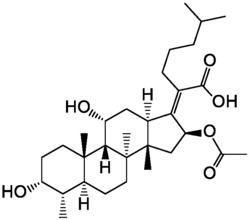
1.式(1)所示的去甲万古霉素类二聚体衍生物:
其中,桥链X为:
2.如权利要求1所述的去甲万古霉素类二聚体衍生物在制备抗菌药物中的用途。
说明书
技术领域
本发明属于医药技术领域,涉及新的糖肽类抗菌素,具体涉及去甲万古霉素类二聚体衍生物、其药学上可接受的盐、及其制备方法,和所述化合物在用于制备治疗或预防细菌感染性疾病的药物中的用途。
背景技术
研究公开了糖肽类抗菌素是一类结构十分复杂的对革兰氏阳性耐药细菌有效的抗菌素,目前临床上应用较多的该类抗菌素包括万古霉素、去甲万古霉素和替考拉宁等;它们在结构上具有相似的七肽氧联的杯状刚性骨架体系和特殊的胺基糖取代基,其中,万古霉素作为具有代表性和应用最广泛的天然糖肽类抗菌素早在1950年代就从土壤放线菌(Amycolatopsis orientalis)的发酵液中分离得到,其抗菌作用机制是与细菌细胞壁肽聚糖前体的三肽序列残基(L-Lys-D-Ala-D-Ala)结合,从而抑制细胞壁的合成;这一特殊的作用机制使得万古霉素与青霉素和头孢菌素等类型的抗菌素不容易发生交叉耐药性,因此在临床上被广泛用于各种耐药性革兰氏阳性菌,特别是耐甲氧西林葡萄球菌(MRSA)、凝固酶阴性葡萄球菌和肠球菌等院内感染的治疗,曾被誉为“抵抗格兰阳性细菌的最后一道防线”。
随着上个世纪80年代首次检测耐万古霉素的肠球菌(VRE)以来,肠球菌对万古霉素的耐药性变得越来越普遍,尤其是近十年来,随着具有高毒力的耐万古霉素金葡菌(VRSA)的出现,使得对耐药细菌所引起的感染性疾病的治疗面临新的重大挑战。因此,发展新一代对耐药革兰氏阳性菌有效的糖肽类抗菌素具有重要意义。在过去的20年中,运用结构修饰的策略制备合成活性万古霉素类似物取得了一些重要进展,2009年9月,FDA批准了第一个用于复杂皮肤和皮下组织感染的万古霉素修饰产物Telavancin,商品名Vibativ;除此之外,另外两个半合成糖肽类衍生物Oritavancin,和Dalbavancin也已经完成III期临床研究,正在进行最后的新药申请阶段。作为第二代糖肽类抗生素的显著代表,上述药物化合物在结构上的特点除了具有七肽氧连的杯状骨架结构之外,在糖基上都具有烷基或芳香基疏水性侧链;构效关系的研究表明,疏水测链的引入是导致糖肽类抗生素对耐药菌恢复抗菌活性的重要原因,抗菌机制研究显示它们不仅仅增加了与细胞壁前体的D-Ala-D-Ala肽残基的亲和力,更可能涉及到对合成细胞壁转糖反应蛋白(转糖酶)的抑制作用,从而达到抑制敏感菌和耐药菌的目的。这种多重抗菌作用机制不仅可以增强对耐药细菌的抑制活性,还可以进一步减少细菌对其新耐药性的产生,从而克服糖肽类抗菌素在临床上广泛应用的主要障碍。
有文献报道万古霉素通过形成共价二聚体,能增强两个正面分别与两个肽聚糖前体末端残基D-Ala-D-Ala结合的亲和力,从而阻断细胞壁肽聚糖的合成,提高对敏感菌和耐药菌的活性。然而,通过去甲万古霉素形成具有抗菌活性的二聚体迄今尚未见有任何文献报道或专利公开。
发明内容
本发明的目地是在克服现有技术的缺陷,提供新的糖肽类抗菌素,具体涉及一种去甲万古霉素类二聚体衍生物、其药学上可接受的盐、及其制备方法,和所述化合物在用于制备治疗或预防细菌感染性疾病的药物中的用途。
本发明的技术方案如下:
本发明提供了通式(1)所示的化合物、其药学上可接受的盐:
其中,R代表氢或取代或非取代的C1-12的烷基、环烷基、杂环烷基、芳香基、杂芳香取代基,以及C1-12的烷基酰基、芳香基酰基和烷基磺酰基;
桥链X为:
上述通式(1)所示的去甲万古霉素衍生物按下式采用下述方法和步骤制备,但本发明化合物的制备方法不限于这些方法。
步骤I:以去甲万古霉素或其衍生物2为原料,加入DIEA和CuSO4
步骤II:将化合物2加入DIEA,N2保护下加入如以下化学式所示的桥链及CuI,TLC监测原料基本反应完全得到化合物1。
本发明的化合物可进一步用于制备药物,尤其是制备抗细菌感染的药物。
本发明中进行了体外抑菌实验,对通式(1)所示的化合物的体外抑菌实验,结果表明,所述化合物对多种格兰氏阳性菌的抑菌活性显著高于万古霉素和去甲万古霉素。
本发明提供了一类新的(去甲)万古霉素衍生物在制备抗菌药物上的应用所述衍生物可用于制备含有所述衍生物作为有效成分的药物。
本发明的(去甲)万古霉素衍生物可制备包含安全有效量新的(去甲)万古霉素衍生物及药用载体的各种制剂,其中的化合物具有比万古霉素或去甲万古霉素更高的抗菌活性。
本发明中,“安全有效量”指的是:化合物的量足以明显改善病情,而不至于产生严重的副作用。安全有效量根据治疗对象的年龄、病情、疗程等来确定。
本发明中,“药用载体”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性;“相容性”在此指的是组合物中各组份能和本发明的化合物以及它们之间相互掺和,而不明显降低化合物的药效;药学上可以接受的载体部分例子有糖(如葡萄糖、蔗糖、乳糖等),淀粉(如玉米淀粉、马铃薯淀粉等),纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等),明胶,滑石,固体润滑剂(如硬脂酸、硬脂酸镁),硫酸钙,植物油(如豆油、芝麻油、花生油、橄榄油等),多元醇(如丙二醇、甘油、甘露醇、山梨醇等),乳化剂(如吐温 )、润湿剂(如十二烷基硫酸钠),着色剂,调味剂,稳定剂,抗氧化剂,防腐剂,无热原水等。
具体实施方式
下面结合实施例对本发明作进一步阐述,但这些实施例绝不是对本发明的任何限制。
实施例1
合成去甲万古霉素二聚体衍生物1a
将先导化合物2a(100mg,0.062mmol)加入25mL三口烧瓶中,用5mL DMF溶解,将温度降至0℃后,加入DIEA(16.03mg,0.124mmol)及0.1%当量的CuSO4.5H2O作为催化剂,氮气保护,0℃下滴加TfN3的DCM溶液到反应体系中,0℃反应8h。TLC监测原料基本反应完全,缓慢滴加DCM至体系中,逐渐有不溶物产生,继续滴加DCM直到不再产生不溶物,离心分离,弃去上清液,得到淡紫色粉末状固体3a(92mg,收率90.7%);
将化合物3a(100mg,0.061mmol)加入10mL三口瓶中,用5mL DMF溶解,加入DIEA(31.6mg,0.244mmol),待化合物3a完全溶解后,N2保护下加入桥链I(8mg,0.0305mmol)及0.1%的CuI,室温反应,HPLC监测原料反应完全,向反应体系中加3-5倍量的EA,离心分离,弃去上清液,固体粗品用MPLC反相柱层析分离纯化,得到目标化合物1a;
ESI-MS m/z:1767.5[M+2H]
根据上述相同的方法,高收率制得了1b所代表的化合物;
ESI-MS m/z:1727.4[M+2H]
根据上述相同的方法,高收率制得了1c所代表的化合物;
ESI-MS m/z:1733.0[M+2H]
实施例2体外抑菌活性的测试
采用实施例1的最终产物(1)a-c进行体外抑菌活性的测试;具体方法按照2006年CLSI(临床实验室标准化协会)推荐的琼脂二倍稀释法进行抗菌药物最低抑菌浓度测定(Minimal Inhibitory concentration MIC)进行;
取不同种类及不同浓度的抗菌药物1ml倒入9cm无菌空平皿中,然后将冷至55℃左右的无菌M-H琼脂19ml立即倾注于平板上,与药液充分混匀,使培养基抗菌药物最终浓度为128、64、32、16、8、4、2、1、0.5、0.25、0.125、0.06μg/ml;同时制备不含抗菌药物的M-H平板作对照;细菌接种将已经孵育18小时的细菌用接种针加入到无菌生理盐水中,配制成浓度为0.5麦氏单位的菌液,然后用无菌盐水稀释10倍,将细菌悬液加入96孔板,根据接种平板的数量,加入不同浓度的菌液;用微量多点接种仪(每点含菌10
表1.化合物的体外抗菌MIC(单位:μg/ml)
其中:金黄色葡萄球菌1102-1106为耐甲氧西林葡萄球菌(MRSA);肠球菌11031-11035、12031-12035为耐万古霉素菌种;肺炎双球菌11061、11062为耐药肺炎球菌菌种。
去甲万古霉素类二聚体衍生物及其制备方法和药用用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0