

IPC分类号 : B01J27/188,B01J27/19,B01J33/00,B01J37/02,B01J37/08,C07C11/18,C07C2/86







专利摘要
本发明提供了一种碳层包覆的复合型催化剂,包括活性组分/SiO2材料以及包覆在所述活性组分/SiO2材料表面的碳层;所述活性组分/SiO2材料由活性组分和SiO2组成,所述活性组分复合在所述SiO2上;所述活性组分包括Cr的化合物、Mo的化合物、W的化合物和P的化合物中的一种或多种。本发明将活性组分负载在载体上,再采用碳层原位包覆,有效的将催化剂活性组分锁住在载体表面,还抑制了由于活性组分和载体之间相互作用力弱,引起的活性组分的脱落的问题,从而有效的提高了催化剂的耐水性,改善了催化剂活性组分的流失率,延长了催化剂的使用寿命,减少了因更换催化剂带来的生产损失,为工业化连续、长期生产提供基础。
权利要求
1.一种用于合成异戊二烯的碳层包覆的复合型催化剂,其特征在于,包括活性组分/SiO
所述活性组分/SiO
所述活性组分包括Cr的化合物、Mo的化合物、W的化合物和P的化合物中的一种或多种;
所述活性组分的质量占所述复合型催化剂总质量的比例为0.1%~30%;
所述碳层的质量占所述复合型催化剂总质量的比例为0.1%~15%;
所述碳层包括无定型碳层和/或石墨化碳层。
2.根据权利要求1所述的复合型催化剂,其特征在于,所述SiO
3.根据权利要求1所述的复合型催化剂,其特征在于,所述活性组分包括Cr的化合物、Mo的化合物、W的化合物和P的化合物中的两种或多种;
所述SiO
所述SiO
所述SiO
4.根据权利要求3所述的复合型催化剂,其特征在于,所述活性组分中,按质量分数计,
所述Cr的化合物为 10~20重量份;
所述P的化合物为5~20重量份;
所述Mo的化合物为 0~20重量份;
所述W的化合物为0~20重量份。
5.根据权利要求1~4任意一项所述的复合型催化剂,其特征在于,所述活性组分还包括稀土元素化合物;
所述稀土元素为La、Ce、Gd和Pr中的一种或多种;
所述稀土元素化合物占所述活性组分的质量比例为0.01%~5%;
所述化合物包括氧化物、氯化物、硝酸盐、硫酸盐、醋酸盐和铵盐中的一种或多种。
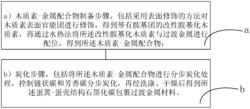
6.一种如权利要求1~5任意一项所述的碳层包覆的复合型催化剂的制备方法,其特征在于,包括以下步骤:
1)将二氧化硅颗粒浸渍到活性组分溶液中,得到催化剂前驱体;
2)将上述步骤得到的催化剂前驱体经过陈化和焙烧后,得到初期催化剂;
3)将碳源包覆在上述步骤得到的初期催化剂上后,得到碳层包覆的复合型催化剂。
7.根据权利要求6所述的制备方法,其特征在于,所述陈化后还包括干燥步骤;
所述浸渍的时间为0.5~4h;
所述陈化的温度为15~30℃;所述陈化的时间为12~36h;
所述干燥的温度为70~120℃;所述干燥的时间为8~19h;
所述焙烧的温度为400~600℃;所述焙烧的时间为3~8h 。
8.根据权利要求7所述的制备方法,其特征在于,所述干燥为梯度升温干燥;
所述步骤3)具体为:
A)在催化剂的作用下,将上述步骤得到的初期催化剂和小分子碳源溶液混合后,原位反应,再在保护性气氛下,碳化后,得到碳层包覆的复合型催化剂;
或
B)在保护性气氛下,通入气体碳源,在上述步骤得到的初期催化剂上原位生长后,得到碳层包覆的复合型催化剂;
或
C)将上述步骤得到的初期催化剂和聚合物碳源溶液混合后,在保护性气氛下,碳化后,得到碳层包覆的复合型催化剂。
9.根据权利要求8所述的制备方法,其特征在于,所述小分子碳源包括糠醇、蔗糖和葡萄糖中的一种或多种;所述气体碳源包括乙烯和/或丁烯;所述聚合物碳源包括聚苯乙烯和/或聚乙烯吡啶;
所述原位反应的温度为50~90℃;所述原位反应的时间为6~36h;
所述原位生长的温度为550~750℃;所述原位生长的时间为0.5~5h;
所述碳化的温度为550~750℃;所述碳化的时间为0.5~10h。
10.权利要求1~5任意一项所述的碳层包覆的复合型催化剂或权利要求6~9任意一项所述的制备方法所制备的碳层包覆的复合型催化剂在催化合成异戊二烯方面的应用。
说明书
技术领域
本发明属于金属催化剂技术领域,涉及一种复合型催化剂及其制备方法、应用,尤其涉及一种碳层包覆的复合型催化剂及其制备方法、在催化合成异戊二烯中的应用。
背景技术
异戊二烯是一种无色易挥发有机化工原料,具有共轭双键结构,主要用于合成橡胶和精细化工产品领域,是人工合成天然橡胶不可取代的烯烃单体,其制备的胶种具有优异的性能,尤其在特殊制品方面的应用,是其他合成橡胶无法企及的。目前异戊二烯主要的制备方法分为物理分离法和化学合成法。物理分离法是最早生产异戊二烯的方法,通过石脑油裂解制乙烯萃取其碳五馏分,随着石油资源的匮乏以及新的乙烯工艺开发,物理分离法制备异戊二烯受到很大制约。特别是近年来,随着橡胶工业的不断发展,对高性能橡胶的需求量日益剧增,这对异戊二烯单体的来源供给提出了巨大挑战,因而发展化学法合成异戊二烯为解决这一难题提供了新思路,其主要分为异丁烯-甲醛法、乙炔丙酮法和丙烯二聚法,其中利用C4资源和甲醛气相一步法工艺简单、投资小、原料成本相对较低的优点,具有可观的经济效益。气相一步法是指以甲醛和异丁烯为原料,在200~400℃常压下,经过缩合脱水直接合成出异戊二烯,流程短、产物易分离、操作简单,在业内受到广泛关注。
因而,发展高效催化剂是气相一步法合成异戊二烯的技术关键,迄今为止,所涉及的催化剂主要有磷系催化剂、铜系催化剂、分子筛催化剂、银系催化剂等,其中磷系催化剂研究较多,也具有一定的竞争优势。中国科学院兰州化学物理研究所与吉林化工研究院共同研制出铬-磷催化剂,通过浸渍法将活性组分负载在SiO2载体上,甲醛的转化率不低于80%,异丁烯选择性不低于61%以上,小试和中试均表现出良好的稳定性。此外,还有银-磷催化剂、硼-磷催化剂、钒-磷催化剂。美国专利US3253051公开了一种Cr、Mn和Ag等金属化合物和磷化物复配的催化剂,这些催化剂都具有较好的工业应用前景。
但是研究表明,上述这些催化剂,在合成异戊二烯过程中,初次使用都可以具有较好的催化性能,但是多次使用后,催化剂的活性均会出现不同程度的衰减,使用寿命较短。
因此,如何延长催化剂的寿命,增强催化剂的长期稳定性,克服上述缺陷,已成为业内诸多具有前瞻性的研究人员广为关注的焦点之一。
发明内容
有鉴于此,本发明要解决的技术问题在于提供一种复合型催化剂及其制备方法,特别是一种碳层包覆的复合型催化剂及其制备方法。本发明制备的碳层包覆的复合型催化剂,在催化合成异戊二烯中,减少了活性组分的流失率,延长了催化剂的使用寿命,同时在催化合成异戊二烯过程中也具有较好的原料转化率和产物收率,有利于工业化生产及推广应用。
本发明提供了一种碳层包覆的复合型催化剂,包括活性组分/SiO2材料以及包覆在所述活性组分/SiO2材料表面的碳层;
所述活性组分/SiO2材料由活性组分和SiO2组成,所述活性组分复合在所述SiO2上;
所述活性组分包括Cr的化合物、Mo的化合物、W的化合物和P的化合物中的一种或多种。
优选的,所述活性组分的质量占所述复合型催化剂总质量的比例为0.1%~30%;
所述碳层的质量占所述复合型催化剂总质量的比例为0.1%~15%;
所述SiO2为SiO2微球;
所述碳层包括无定型碳层和/或石墨化碳层。
优选的,所述活性组分包括Cr的化合物、Mo的化合物、W的化合物和P的化合物中的两种或多种;
所述SiO2的比表面积为100~400m2/g;
所述SiO2的孔径为5~80nm;
所述SiO2的粒径为50~500μm。
优选的,所述活性组分中,按质量分数计,
所述Cr的化合物为 10~20重量份;
所述P的化合物为 5~20重量份;
所述Mo的化合物为 0~20重量份;
所述W的化合物为 0~20重量份。
优选的,所述活性组分还包括稀土元素化合物;
所述稀土元素为La、Ce、Gd和Pr中的一种或多种;
所述稀土元素化合物占所述活性组分的质量比例为0.01%~5%;
所述化合物包括氧化物、氯化物、硝酸盐、硫酸盐、醋酸盐和铵盐中的一种或多种。
本发明提供了一种如上述技术方案任意一项所述的碳层包覆的复合型催化剂的制备方法,包括以下步骤:
1)将二氧化硅颗粒浸渍到活性组分溶液中,得到催化剂前驱体;
2)将上述步骤得到的催化剂前驱体经过陈化和焙烧后,得到初期催化剂;
3)将碳源包覆在上述步骤得到的初期催化剂上后,得到碳层包覆的复合型催化剂。
优选的,所述陈化后还包括干燥步骤;
所述浸渍的时间为0.5~4h;
所述陈化的温度为15~30℃;所述陈化的时间为12~36h;
所述干燥的温度为70~120℃;所述干燥的时间为8~19h;
所述焙烧的温度为400~600℃;所述焙烧的时间为3~8h。
优选的,所述干燥为梯度升温干燥;
所述步骤3)具体为:
A)在催化剂的作用下,将上述步骤得到的初期催化剂和小分子碳源溶液混合后,原位反应,再在保护性气氛下,碳化后,得到碳层包覆的复合型催化剂;
或
B)在保护性气氛下,通入气体碳源,在上述步骤得到的初期催化剂上原位生长后,得到碳层包覆的复合型催化剂;
或
C)将上述步骤得到的初期催化剂和聚合物碳源溶液混合后,在保护性气氛下,碳化后,得到碳层包覆的复合型催化剂。
优选的,所述小分子碳源包括糠醇、蔗糖和葡萄糖中的一种或多种;所述气体碳源包括乙烯和/或丁烯;所述聚合物碳源包括聚苯乙烯和/或聚乙烯吡啶;
所述原位反应的温度为50~90℃;所述原位反应的时间为6~36h;
所述原位生长的温度为550~750℃;所述原位生长的时间为0.5~5h;
所述碳化的温度为550~750℃;所述碳化的时间为0.5~10h。
本发明还提供了上述技术方案任意一项所述的碳层包覆的复合型催化剂或上述技术方案任意一项所述的制备方法所制备的碳层包覆的复合型催化剂在催化合成异戊二烯方面的应用。
本发明提供了一种碳层包覆的复合型催化剂,包括活性组分/SiO2材料以及包覆在所述活性组分/SiO2材料表面的碳层;所述活性组分/SiO2材料由活性组分和SiO2组成,所述活性组分复合在所述SiO2上;所述活性组分包括Cr的化合物、Mo的化合物、W的化合物和P的化合物中的一种或多种。本发明提供了一种碳层包覆的复合型催化剂的制备方法。与现有技术相比,本发明针对现有的金属化合物或复配的催化剂存在使用寿命短的问题,经过研究实验,发现在合成异戊二烯过程中,现有此类催化剂的活性组分严重流失,连续使用50h组分流失高于36wt%。本发明又从该问题入手,认为该合成过程中以福尔马林溶液为甲醛来源,涉及大量的水蒸汽,且催化剂为负载型,载体与活性组分间作用力不强,导致活性组分不断流失,从而对工业化带来了极大的不便和损失。
本发明针对上述原因,创造性的从催化剂的结构方面入手,再选择特定的活性组分,得到了碳层包覆的复合型催化剂,是一种高活性、耐水抗流失的催化剂。本发明将活性组分负载在载体上,再采用碳层原位包覆,有效的将催化剂活性组分锁住在载体表面;另一方面,还抑制了由于活性组分和载体之间相互作用力弱,引起的活性组分的脱落的问题,从而有效的降低了催化剂活性组分在反应过程的中流失。本发明提供的催化剂有效的提高了催化剂的耐水性,改善了催化剂活性组分的流失率,从而延长了催化剂的使用寿命,减少了因更换催化剂带来的生产损失,为工业化连续、长期生产提供基础。
实验结果表明,本发明提供的催化剂具有高活性、耐水抗流失的特性。在同等条件下,连续使用50h后,甲醛转化率不低于66%,选择性不低于78%,异丁烯选择性保持80%以上,活性组分的流失率比同等条件下普通催化剂减少了10%~24%。
附图说明
图1为本发明实施例1制备的碳层包覆的复合型催化剂和未包覆碳层的初期催化剂的外观照片;
图2为本发明实施例11制备的催化剂和普通催化剂在催化过程中的活性组分流失速率曲线。
具体实施方式
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为了进一步说明本发明的特征和优点,而不是对发明权利要求的限制。
本发明所有原料,对其来源没有特别限制,在市场上购买的或按照本领域技术人员熟知的常规方法制备的即可。
本发明所有原料,对其纯度没有特别限制,本发明优选采用分析纯或原子层沉积领域常规的纯度要求。
本发明所有原料和工艺过程,其牌号或简称均属于本领域常规牌号或简称,每个牌号或简称在其相关用途的领域内均是清楚明确的,本领域技术人员根据牌号、简称以及相应的用途,能够从市售中购买得到或常规方法制备得到,或者采用相应的设备进行实现。
本发明提供了一种碳层包覆的复合型催化剂,包括活性组分/SiO2材料以及包覆在所述活性组分/SiO2材料表面的碳层;
所述活性组分/SiO2材料由活性组分和SiO2组成,所述活性组分复合在所述SiO2上;
所述活性组分包括Cr的化合物、Mo的化合物、W的化合物和P的化合物中的一种或多种。
本发明对所述碳层的厚度没有特别限制,以本领域技术人员熟知的常规核壳型结构复合物的层厚度即可,本领域技术人员可以根据实际应用需要、产品要求以及质量要求进行选择和调整。本发明对所述碳层的形式没有特别限制,以本领域技术人员熟知的常规碳层的形式即可,本领域技术人员可以根据实际应用需要、产品要求以及质量要求进行选择和调整,本发明所述碳层的形式优选包括无定型碳层和/或石墨化碳层,更优选为无定型碳层和石墨化碳层。本发明对所述碳层的比例没有特别限制,以本领域技术人员熟知的常规核壳型结构复合物的层比例即可,本领域技术人员可以根据实际应用需要、产品要求以及质量要求进行选择和调整,本发明为进一步保证产品的催化寿命,所述碳层的质量占所述复合型催化剂总质量的比例优选为0.1%~15%,更优选为0.5%~13%,更优选为1%~10%,更优选为4%~7%。
本发明中活性组分/SiO2材料由活性组分和SiO2组成。所述活性组分复合在所述SiO2上。
本发明对所述复合的定义没有特别限制,以本领域技术人员熟知的常规复合的定义即可,本领域技术人员可以根据实际应用需要、产品要求以及质量要求进行选择和调整,本发明为进一步保证产品的催化性能,所述复合优选包括负载、嵌入、生长、包覆、掺杂、吸附和键连中的一种或多种,更优选为负载、嵌入、生长、包覆、掺杂、吸附或键连,最优选为负载,具体可以为均匀负载和/或致密负载。本发明中复合在所述SiO2上优选包括复合在SiO2表面和/或其内部孔道中,更优选为复合在SiO2表面和其内部孔道中。
本发明对所述SiO2的形态和参数没有特别限制,以本领域技术人员熟知的常规二氧化硅材料的形态和参数即可,本领域技术人员可以根据实际应用需要、产品要求以及质量要求进行选择和调整,本发明为进一步保证产品的催化性能,所述SiO2在形貌上,优选为SiO2微球。本发明所述SiO2的粒径优选为50~500μm,更优选为100~400μm,更优选为200~300μm。本发明所述SiO2的比表面积优选为100~400m2/g,更优选为150~350m2/g,更优选为200~300m2/g。本发明所述SiO2的孔径优选为5~80nm,更优选为10~70nm,更优选为20~60nm,更优选为30~50nm。
本发明对所述活性组分的用量没有特别限制,以本领域技术人员熟知的常规此类催化剂的常规用量即可,本领域技术人员可以根据实际应用需要、产品要求以及质量要求进行选择和调整,本发明为进一步保证产品的催化性能,所述活性组分的质量占所述复合型催化剂总质量的比例优选为0.1%~30%,更优选为1%~25%,更优选为5%~20%,更优选为10%~15%,具体可以为15%~25%。
本发明所述活性组分包括Cr的化合物、Mo的化合物、W的化合物和P的化合物中的一种或多种,更优选为Cr的化合物、Mo的化合物、W的化合物和P的化合物中的两种或多种,更优选为Cr的化合物和P的化合物,或者,Cr的化合物、P的化合物以及另外两种化合物中的一种和两种。
本发明对所述活性组分为上述多个化合物时的具体比例没有特别限制,本领域技术人员可以根据实际应用需要、产品要求以及质量要求进行选择和调整,本发明所述活性组分中,按质量分数计,所述Cr的化合物优选为10~20重量份;所述P的化合物优选为5~20重量份;所述Mo的化合物优选为0~20重量份;所述W的化合物优选为0~20重量份。其中,所述Cr的化合物更优选为12~18重量份,更优选为14~16重量份。所述P的化合物更优选为7~18重量份,更优选为10~15重量份。所述Mo的化合物更优选为3~18重量份,更优选为5~15重量份,更优选为8~13重量份。所述W的化合物更优选为3~18重量份,更优选为5~15重量份,更优选为8~13重量份。
本发明为进一步提高活性组分的活性和催化剂的性能,延长催化剂的使用寿命,所述活性组分优选还包括稀土元素化合物;所述稀土元素为La、Ce、Gd和Pr中的一种或多种,更优选为La、Ce、Gd或Pr。本发明对所述稀土元素化合物的具体用量没有特别限制,本领域技术人员可以根据实际应用需要、产品要求以及质量要求进行选择和调整,本发明所述稀土元素化合物占所述活性组分的质量比例优选为0.01%~5%,更优选为0.1%~4%,更优选为0.5%~3%,更优选为1%~2%。
本发明对前述所有元素的化合物中,具体化合物的选择没有特别限制,以本领域技术人员熟知的上述元素的常规化合物即可,本领域技术人员可以根据实际生产需要、产品要求以及质量要求进行选择和调整,本发明所述化合物优选包括可溶性化合物,更优选包括氧化物、氯化物、硝酸盐、硫酸盐、醋酸盐和铵盐中的一种或多种,更优选为氧化物、氯化物、硝酸盐、硫酸盐、醋酸盐或铵盐,最优选为氧化物。在本发明中,前述各化合物的质量比例优选为按其氧化物的比例进行设定。
具体的,本发明所述化合物具体为:
所述Cr的化合物可以为Cr2O3、CrO3、Cr(NO3)3或CrCl3·6H2O。所述Mo的化合物可以为MoO3、MoCl5、(NH4)6Mo7O24或H3PMo12O40·xH2O。所述W的化合物可以为WO3、(NH4)2WO4、(NH4)10W12O41·xH2O或H3PW12O40·xH2O。所述P的化合物可以为H3PO4、P2O5、(NH4)3PO4、(NH4)2HPO4或NH4H2PO4。所述稀土La、Ce、Gd、Pr的化合物可以为La(NO3)3·6H2O、La2O3、Ce(NO3)3·6H2O、CeCl3、Gd(NO3)3·6H2O或Pr(NO3)3·6H2O。
本发明还提供了一种如上述技术方案任意一项所述的碳层包覆的复合型催化剂的制备方法,其特征在于,包括以下步骤:
1)将二氧化硅颗粒浸渍到活性组分溶液中,得到催化剂前驱体;
2)将上述步骤得到的催化剂前驱体经过陈化和焙烧后,得到初期催化剂;
3)将碳源包覆在上述步骤得到的初期催化剂上后,得到碳层包覆的复合型催化剂。
本发明对上述制备方法中碳层包覆的复合型催化剂的元素选择和比例,以及相应的优选原则,与前述碳层包覆的复合型催化剂中所对应元素的选择和比例,以及相应的优选原则可以进行对应,在此不再一一赘述。
本发明首先将二氧化硅颗粒浸渍到活性组分溶液中,得到催化剂前驱体。
本发明对所述浸渍的条件没有特别限制,以本领域技术人员熟知的此类浸渍的常规参数即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明为提高最终产品的性能,所述浸渍优选为等体积浸渍。本发明所述浸渍的温度优选常温浸渍,更优选为0~40℃,更优选为5~35℃,更优选为10~25℃。本发明所述浸渍的时间优选为0.5~4h,更优选为1~3.5h,更优选为1.5~3h,更优选为2~2.5h。
本发明对所述活性组分溶液的溶剂选择没有特别限制,以本领域技术人员熟知的此类化合物的常规溶剂即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明所述溶剂优选为水和/或乙醇,更优选为水。
本发明随后将上述步骤得到的催化剂前驱体经过陈化和焙烧后,得到初期催化剂。
本发明对所述陈化的方式没有特别限制,以本领域技术人员熟知的常规陈化方式即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明所述陈化的方式优选为静置。本发明对所述陈化的参数没有特别限制,以本领域技术人员熟知的常规陈化参数即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明为提高最终产品的性能,所述陈化的温度优选为15~30℃,更优选为17~28℃,更优选为20~25℃。本发明所述陈化的时间优选为12~36h,更优选为18~30h,更优选为21~27h。
本发明对所述焙烧的参数没有特别限制,以本领域技术人员熟知的常规焙烧参数即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明为提高最终产品的性能,所述焙烧的温度优选为400~600℃,更优选为420~580℃,更优选为450~550℃。本发明所述焙烧的时间优选为3~8h,更优选为4~7h,更优选为5~6h。
本发明为进一步提高活性组分的活性和催化剂的性能,延长催化剂的使用寿命,所述陈化之后,即陈化和焙烧之间,还包括干燥步骤。
本发明对所述干燥的参数没有特别限制,以本领域技术人员熟知的常规干燥参数即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明为提高最终产品的性能,所述干燥的温度优选为70~120℃,更优选为80~110℃,更优选为90~100℃。本发明所述干燥的时间优选为8~19h,更优选为10~17h,更优选为12~15h。
本发明对所述干燥的方式没有特别限制,以本领域技术人员熟知的常规干燥方式即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明为提高最终产品的性能,所述干燥的方式优选为梯度升温干燥。具体过程可以为:
先在65~75℃(更优选为67~73℃,更优选为69~71℃)温度下干燥1~3h(更优选为1.5~2.5h),然后在80~90℃(更优选为82~88℃,更优选为84~86℃)温度下干燥1~3h(更优选为1.5~2.5h),再在100~110℃(更优选为102~108℃,更优选为104~106℃)温度下干燥1~3h(更优选为1.5~2.5h),最后在115~125℃(更优选为117~123℃,更优选为119~121℃)温度下干燥5~10h(更优选为6~9h,更优选为7~8h)。
本发明最后将碳源包覆在上述步骤得到的初期催化剂上后,得到碳层包覆的复合型催化剂。
本发明对所述碳源没有特别限制,以本领域技术人员熟知的常规包覆用碳源前驱体即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明所述碳源优选包括富碳小分子、大分子,气体、液体或聚合物树脂。
本发明对所述包覆的具体方式没有特别限制,以本领域技术人员熟知的常规包覆方式即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明为保证产品的催化性能以及使用寿命,所述包覆的方式,即步骤3)优选包括以下三种方式:
A)在催化剂的作用下,将上述步骤得到的初期催化剂和小分子碳源溶液混合后,原位反应,再在保护性气氛下,碳化后,得到碳层包覆的复合型催化剂;
或
B)在保护性气氛下,通入气体碳源,在上述步骤得到的初期催化剂上原位生长后,得到碳层包覆的复合型催化剂;
或
C)将上述步骤得到的初期催化剂和聚合物碳源溶液混合后,在保护性气氛下,碳化后,得到碳层包覆的复合型催化剂。
其中,A)在催化剂的作用下,将上述步骤得到的初期催化剂和小分子碳源溶液混合后,原位反应,再在保护性气氛下,碳化后,得到碳层包覆的复合型催化剂。
本发明所述催化剂优选包括草酸。本发明所述小分子碳源优选包括糠醇、蔗糖和葡萄糖中的一种或多种,更优选为糠醇、蔗糖或葡萄糖,最优选为糠醇。本发明所述小分子碳源溶液的质量浓度优选为1%~80%,更优选为10%~70%,更优选为30%~50%。本发明所述小分子碳源溶液的溶剂优选包括乙醇、甲苯和三甲苯中的一种或多种,更优选为乙醇、甲苯或三甲苯,最优选为三甲苯。本发明所述保护性气氛优选包括氮气和/或惰性气体,更优选为氮气或氩气。
本发明所述混合的方式优选为浸渍,更优选为等体积浸渍,更优选为多次浸渍。所述浸渍的次数优选为1~5次,更优选为2~4次。本发明所述原位反应的温度优选为50~90℃,更优选为60~80℃,更优选为65~75℃;所述原位反应的时间优选为6~36h,更优选为12~30h,更优选为18~24h,具体可以为12~24h。本发明所述碳化的温度优选为550~750℃,更优选为580~720℃,更优选为600~700℃。所述碳化的时间优选为0.5~10h,更优选为1~8h,更优选为2~7h,更优选为4~6h。
本发明为完整和优化工艺过程,保证最终产品的性能,上述步骤具体可以为:
以液体浸渍液为碳源
用碳源溶液浸渍初期催化剂,在催化剂的作用下,原位反应后再高温碳化,即将液体小分子碳源和交联催化剂通过等体积浸渍的方式覆盖到催化剂表面,可多次浸渍,在50~90℃条件原位反应,惰性气氛中550~750℃下碳化。
更具体优选为:
以液体浸渍液为碳源
(a)配制1~80wt%的碳源溶液,加入草酸做催化剂;
(b)将初期催化剂等体积浸渍在上述碳源溶液中,室温下静置12~36h,50~90℃烘干;
(c)将(b)所得的产物在高纯氩气气氛下550~750℃下保持0.5~10h,得到碳层包覆的复合型催化剂。
其中,B)在保护性气氛下,通入气体碳源,在上述步骤得到的初期催化剂上原位生长后,得到碳层包覆的复合型催化剂。
本发明所述气体碳源优选包括丁烯和/或乙烯,更优选为丁烯或乙烯,最优选为丁烯。本发明所述气体碳源的体积分数优选为20%~90%,更优选为30%~80%,更优选为50%~60%。本发明所述保护性气氛优选包括氮气和/或惰性气体,更优选为氮气或氩气,最优选为氩气。本发明所述原位生长的设备优选为气氛炉。
本发明所述原位生长的温度优选为550~750℃,更优选为580~720℃,更优选为600~700℃。所述原位生长的时间优选为0.5~5h,更优选为1~4h,更优选为2~3h。
本发明为完整和优化工艺过程,保证最终产品的性能,上述步骤具体可以为:
以气体分子为碳源
在惰性气氛中550~750℃下通入气体碳源,在初期催化剂上原位生成碳层
更具体优选为:
以气体分子为碳源
(a)将初期催化剂放置于气氛炉中,通入氩气;
(b)升温至550℃~750℃下,通入碳源气体,其中碳源气体体积分数为20~90vol%,通入碳源的时间为0.5~5h;
(c)结束后关闭碳源气体,保持氩气气氛至系统温度降至室温。
其中,C)将上述步骤得到的初期催化剂和聚合物碳源溶液混合后,在保护性气氛下,碳化后,得到碳层包覆的复合型催化剂。
本发明所述聚合物碳源优选包括聚苯乙烯和/或聚乙烯吡啶,更优选为聚苯乙烯或聚乙烯吡啶,最优选为聚苯乙烯。本发明所述聚合物碳源溶液的质量浓度优选为1%~30%,更优选为5%~25%,更优选为10%~20%。本发明所述聚合物碳源溶液的溶剂优选包括甲苯、氯仿、乙醇和丙酮中的一种或多种,更优选为甲苯、氯仿、乙醇或丙酮,最优选为甲苯。本发明所述保护性气氛优选包括氮气和/或惰性气体,更优选为氮气或氩气。
本发明所述混合的方式优选为浸渍,更优选为等体积浸渍,更优选为多次浸渍。所述浸渍的次数优选为1~5次,更优选为2~4次。本发明所述碳化的温度优选为550~750℃,更优选为580~720℃,更优选为600~700℃。所述碳化的时间优选为0.5~10h,更优选为1~8h,更优选为2~7h,更优选为4~6h。
本发明为完整和优化工艺过程,保证最终产品的性能,上述步骤具体可以为:
以液体浸渍液为碳源
用聚合物溶液浸渍初期催化剂,通过等体积浸渍的方式覆盖到催化剂表面,可多次浸渍,在惰性气氛中550~750℃下碳化。
更具体优选为:
以液体浸渍液为碳源
(a)将聚合物溶解,配成1~30wt%的溶液;
(b)将初期催化剂用上述溶液等体积浸渍1~5次,干燥;
(c)将(b)所得的产物在氩气气氛下,550℃~750℃保持0.5~10h,得到碳层包覆的复合型催化剂。
本发明上述步骤提供了一种碳层包覆的复合型催化剂的制备方法,为完整和细化具体工艺,上述步骤具体可以为:
第一步,将活性组分按照设计量溶解在水中,加入载体SiO2微球浸渍得到催化剂前驱体;
第二步,将催化剂前驱体陈化、梯度升温干燥、焙烧,得到初期催化剂;
第三步,将初期催化剂用碳层包覆,碳层通过碳源在催化剂表面原位生长。
本发明还提供了上述技术方案任意一项所述的碳层包覆的复合型催化剂或上述技术方案任意一项所述的制备方法所制备的碳层包覆的复合型催化剂在催化合成异戊二烯方面的应用。
本发明对所述异戊二烯的具体制备过程没有特别限制,以本领域技术人员熟知的异戊二烯的制备方法即可,本领域技术人员可以根据实际生产情况、产品要求以及质量要求进行选择和调整,本发明所述异戊二烯的制备方法优选为气相一步法合成异戊二烯,更优选为烯醛气相一步法合成异戊二烯,具体过程可以为,常压下,在250℃氮气条件下,保持烯醛比为4:1,催化剂接触时间为1.0s的反应条件下,进行合成。
本发明上述步骤提供了一种碳层包覆的复合型催化剂及其制备方法、应用,本发明从催化剂的结构方面入手,再选择特定的活性组分,得到了碳层包覆的复合型催化剂,是一种用于气相法合成异戊二烯的高活性、耐水抗流失的催化剂。本发明将碳层原位包覆,有效的锁住催化剂的活性组分;另一方面,还抑制了由于活性组分和载体之间相互作用力弱,引起的活性组分的脱落的问题,从而有效的降低了催化剂活性组分在反应过程的中流失。
本发明提供的催化剂由活性组分、载体和碳包覆层组成,通过特定的制备方法及过程参数,采用浸渍法将活性组分负载在载体上,并通过原位生长的碳层包覆,将催化剂活性组分锁住在载体表面,有效的提高了催化剂的耐水性,改善了催化剂活性组分的流失率,从而延长了催化剂的使用寿命,减少了因更换催化剂带来的生产损失,为工业化连续、长期生产提供基础。
实验结果表明,本发明提供的催化剂具有高活性、耐水抗流失的特性。在同等条件下,连续使用50h后,甲醛转化率不低于66%,选择性不低于78%,异丁烯选择性保持80%以上,活性组分的流失率比同等条件下普通催化剂减少了10%~24%。
为了进一步说明本发明,以下结合实施例对本发明提供的一种复合型催化剂及其制备方法、应用进行详细描述,但是应当理解,这些实施例是在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制,本发明的保护范围也不限于下述的实施例。
实施例1
(1)取1.50g CrO3,0.61g钼酸铵和1.63g磷酸溶于13ml去离子水中,配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体20℃下陈化12h;然后梯度升温干燥,70℃保持1.5h,85℃保持1.5h,105℃保持1.5h,120℃保持5h;最后500℃下焙烧5h,得到初期催化剂;
(3)以三甲苯为溶剂,配制40wt%的糠醇溶液,加入微量的草酸作为催化剂,将(2)中所得的初期催化剂用糠醇溶液等体积浸渍2次,50℃干燥10h,90℃干燥6h;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至550℃,保持5h,结束后氩气气氛下冷却至室温,得到催化剂,即碳层包覆的复合型催化剂。
对本发明实施例1制备的碳层包覆的复合型催化剂进行表征。
参见图1,图1为本发明实施例1制备的碳层包覆的复合型催化剂和未包覆碳层的初期催化剂的外观照片。
其中,(a)为SiO2负载的初期催化剂,(b)包覆碳层的复合催化剂。
由图1可知,本发明制备得到了碳层包覆的复合型催化剂,在活性组分/SiO2材料的表面包覆了碳层。
对本发明实施例1制备的碳层包覆的复合型催化剂进行热失重分析。
由热失重分析结果可知,在空气条件下,对比初期催化剂,碳层包覆的复合催化剂从350℃开始失重,随着温度升高,质量减少明显,从而表明,本发明得到了碳层包覆的复合型催化剂。
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为89%,甲醛选择性为54%,异丁烯选择性为79%,反应50h后,甲醛转化率为67%,选择性为81%,异丁烯选择性为90%,同比条件下催化剂活性组分流失率减少18%。
由此可以看出,本发明提供的碳层包覆的复合型催化剂明显延长了催化剂的使用寿命,而且随着使用的时间,还能够大大提升原料和产品的选择性。
实施例2
(1)取1.20g Cr2O3,0.24g钼酸铵和1.63g磷酸溶于13ml去离子水中,配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体25℃下陈化24h;然后梯度升温干燥,70℃保持1.5h,85℃保持1.5h,105℃保持1.5h,120℃保持5h;最后550℃下焙烧5h,得到初期催化剂;
(3)以三甲苯为溶剂,配制20wt%的糠醇溶液,加入微量的草酸作为催化剂,将(2)中所得的初期催化剂用糠醇溶液等体积浸渍3次,60℃干燥6h,80℃干燥6h;;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至650℃,保持5h,结束后氩气气氛下冷却至室温,得到催化剂;
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为84%,反应50h后,甲醛转化率为66%,选择性为79%,异丁烯选择性为85%,同比条件下催化剂活性组分流失率减少15%。
实施例3
(1)取1.00g CrO3,1.20g钼酸铵和2.27g磷酸铵溶于13ml去离子水中,加入0.02g硝酸镧,配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体20℃下陈化12h;然后梯度升温干燥,70℃保持1.5h,85℃保持1.5h,105℃保持1.5h,120℃保持5h;最后500℃下焙烧5h,得到初期催化剂;
(3)以三甲苯为溶剂,配制80wt%的糠醇溶液,加入微量的草酸作为催化剂,将(2)中所得的初期催化剂用糠醇溶液等体积浸渍1次,70℃干燥8h,80℃干燥8h;;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至650℃,保持5h,结束后氩气气氛下冷却至室温,得到催化剂;
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为89%,反应50h后,甲醛转化率为70%,选择性为83%,异丁烯选择性为87%,同比条件下催化剂活性组分流失率减少15%。
实施例4
(1)取1.00g CrO3,0.61g钼酸铵和1.63g磷酸溶于13ml去离子水中,加入0.02g硝酸铈和0.40g磷钨酸,配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体30℃下陈化24h;然后梯度升温干燥,70℃保持2h,85℃保持12h,105℃保持2h,120℃保持4h;最后550℃下焙烧8h,得到初期催化剂;
(3)以三甲苯为溶剂,配制40wt%的糠醇溶液,加入微量的草酸作为催化剂,将(2)中所得的初期催化剂用糠醇溶液等体积浸渍2次,60℃干燥10h,90℃干燥5h;;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至650℃,保持3h,结束后氩气气氛下冷却至室温,得到催化剂;
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为86%,反应50h后,甲醛转化率为71%,选择性为83%,异丁烯选择性为85%,同比条件下催化剂活性组分流失率减少16%。
实施例5
(1)取1.20g CrO3,0.4g磷钨酸和2.43g磷酸溶于13ml去离子水中,加入0.02g硝酸钆配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体30℃下陈化12h;然后梯度升温干燥,70℃保持1.5h,85℃保持1.5h,105℃保持1.5h,120℃保持8h;最后600℃下焙烧3h,得到初期催化剂;
(3)以丁烯为碳源,将(2)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至550℃,通入丁烯气体,保持丁烯体积分数站总气体体积的50vol%,保持3h,结束后关闭丁烯,继续通入氩气条件下冷却至室温,得到催化剂;
(4)将(3)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为88%,反应50h后,甲醛转化率为68%,选择性为81%,异丁烯选择性为82%,同比条件下催化剂活性组分流失率减少10%。
实施例6
(1)取1.50g CrO3,0.61g钼酸铵和2.43g磷酸溶于13ml去离子水中;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体25℃下陈化12h;然后梯度升温干燥,70℃保持2h,85℃保持2h,105℃保持2h,120℃保持3h;最后550℃下焙烧5h,得到初期催化剂;
(3)以丁烯为碳源,将(2)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至650℃,通入丁烯气体,保持丁烯体积分数站总气体体积的80vol%,保持1h,结束后关闭丁烯,继续通入氩气条件下冷却至室温,得到催化剂;
(4)将(3)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为86%,反应50h后,甲醛转化率为69%,选择性为79%,异丁烯选择性为81%,同比条件下催化剂活性组分流失率减少12%。
实施例7
(1)取1.00g CrO3,0.61g钼酸铵和2.27g磷酸溶于13ml去离子水中,加入0.04g硝酸钆配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体20℃下陈化24h;然后梯度升温干燥,70℃保持1h,85℃保持1h,105℃保持1h,120℃保持8h;最后600℃下焙烧3h,得到初期催化剂;
(3)以丁烯为碳源,将(2)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至600℃,通入丁烯气体,保持丁烯体积分数站总气体体积的30vol%,保持5h,结束后关闭丁烯,继续通入氩气条件下冷却至室温,得到催化剂;
(4)将(3)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为90%,反应50h后,甲醛转化率为70%,选择性为82%,异丁烯选择性为85%,同比条件下催化剂活性组分流失率减少10%。
实施例8
(1)取1.20g CrO3,0.80g磷钨酸和2.43g磷酸溶于13ml去离子水中,加入0.04g硝酸镨配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体20℃下陈化36h;然后梯度升温干燥,70℃保持2h,85℃保持2h,105℃保持2h,120℃保持3h;最后550℃下焙烧8h,得到初期催化剂;
(3)以丁烯为碳源,将(2)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至550℃,通入丁烯气体,保持丁烯体积分数站总气体体积的80vol%,保持1h,结束后关闭丁烯,继续通入氩气条件下冷却至室温,得到催化剂;
(4)将(3)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为89%,反应50h后,甲醛转化率为70%,选择性为83%,异丁烯选择性为85%,同比条件下催化剂活性组分流失率减少12%。
实施例9
(1)取0.80g CrO3,0.61g钼酸铵和0.82g磷酸溶于13ml去离子水中,加入0.04g磷钨酸和0.04g硝酸钆配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体25℃下陈化24h;然后梯度升温干燥,70℃保持1.5h,85℃保持1.5h,105℃保持1.5h,120℃保持8h;最后550℃下焙烧5h,得到初期催化剂;
(3)以三甲苯为溶剂,配制40wt%的糠醇溶液,加入微量的草酸作为催化剂,将(2)中所得的初期催化剂用糠醇溶液等体积浸渍3次;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至650℃,保持8h,结束后氩气气氛下冷却至室温,得到催化剂;
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为89%,反应50h后,甲醛转化率为69%,选择性为83%,异丁烯选择性为84%,同比条件下催化剂活性组分流失率减少17%。
实施例10
(1)取0.80g CrO3,0.25g钼酸铵和0.81g磷酸溶于13ml去离子水中,加入0.08g磷钨酸和0.04g硝酸镨配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体20℃下陈化12h;然后梯度升温干燥,70℃保持1.5h,85℃保持1.5h,105℃保持1.5h,120℃保持5h;最后550℃下焙烧5h,得到初期催化剂;
(3)以甲苯为溶剂,配制2wt%的聚苯乙烯溶液,将(2)中所得的初期催化剂用聚苯乙烯的甲苯溶液等体积浸渍4次;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至600℃,保持8h,结束后氩气气氛下冷却至室温,得到催化剂;
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为91%,反应50h后,甲醛转化率为71%,选择性为82%,异丁烯选择性为85%,同比条件下催化剂活性组分流失率减少20%。
实施例11
(1)取1.00g CrO3和0.81g磷酸溶于13ml去离子水中,加入1.00g磷钨酸和0.04g硝酸镨配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体25℃下陈化12h;然后梯度升温干燥,70℃保持1h,85℃保持1h,105℃保持1h,120℃保持8h;最后600℃下焙烧3h,得到初期催化剂;
(3)以甲苯为溶剂,配制10wt%的聚苯乙烯溶液,将(2)中所得的初期催化剂用聚苯乙烯的甲苯溶液等体积浸渍1次;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至650℃,保持3h,结束后氩气气氛下冷却至室温,得到催化剂;
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为85%,反应50h后,甲醛转化率为66%,选择性为83%,异丁烯选择性为82%,同比条件下催化剂活性组分流失率减少24%。
参见图2,图2为本发明实施例11制备的催化剂和普通催化剂在催化过程中的活性组分流失速率曲线。
由图2可知,本发明制备的催化剂有效的提高了催化剂的耐水性,大大降低了催化剂活性组分的流失率,从而延长了催化剂的使用寿命。
实施例12
(1)取1.20g CrO3,1.20g钼酸铵和1.63g磷酸溶于13ml去离子水中,配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体25℃下陈化24h;然后梯度升温干燥,70℃保持1.5h,85℃保持1.5h,105℃保持1.5h,120℃保持5h;最后550℃下焙烧8h,得到初期催化剂;
(3)以甲苯为溶剂,配制5wt%的聚苯乙烯溶液,将(2)中所得的初期催化剂用聚苯乙烯的甲苯溶液等体积浸渍4次;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至600℃,保持5h,结束后氩气气氛下冷却至室温,得到催化剂;
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为86%,反应50h后,甲醛转化率为67%,选择性为80%,异丁烯选择性为84%,同比条件下催化剂活性组分流失率减少19%。
实施例13
(1)取1.00g CrO3,0.61g钼酸铵和1.63g磷酸溶于13ml去离子水中,加入0.8g磷钨酸和0.04g硝酸钆配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体25℃下陈化24h;然后梯度升温干燥,70℃保持2h,85℃保持2h,105℃保持2h,120℃保持3h;最后550℃下焙烧8h,得到初期催化剂;
(3)以三甲苯为溶剂,配制40wt%的糠醇溶液,加入微量的草酸作为催化剂,将(2)中所得的初期催化剂用糠醇溶液等体积浸渍1次;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至550℃,通入丁烯气体,保持丁烯含量为20vol%,保持5h,结束后关闭丁烯,在氩气气氛下冷却至室温,得到催化剂;
(5)将(4)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为89%,反应50h后,甲醛转化率为70%,选择性为81%,异丁烯选择性为82%,同比条件下催化剂活性组分流失率减少20%。
实施例14
(1)取0.80g CrO3和1.95g磷酸溶于13ml去离子水中,加入0.40g磷钨酸和0.04g硝酸钆配成浸渍液;取10g球形SiO2,将浸渍液加入到SiO2载体上,获得催化剂体前驱体;
(2)将(1)中所述催化剂前驱体20℃下陈化12h;然后梯度升温干燥,70℃保持1.5h,85℃保持1.5h,105℃保持1.5h,120℃保持5h;最后500℃下焙烧8h,得到初期催化剂;
(3)以三甲苯为溶剂,配制40wt%的糠醇溶液,加入微量的草酸作为催化剂,将(2)中所得的初期催化剂用糠醇溶液等体积浸渍1次;
(4)将(3)中所得催化剂置于气氛炉中,高纯氩气保护下,以3℃/min的升温速率升高至550℃,保持3h,结束后氩气气氛下冷却至室温;
(5)将(4)中所得到催化剂用2wt%聚苯乙烯得甲苯溶液等体积浸渍1次;
(6)将(5)中所得催化剂在固定床评价装置上用于合成异戊二烯,烯醛比为4:1,接触时间为1.0s,连续反应50h后,进行检测。
结果表明,反应初期,甲醛转化率为88%,反应50h后,甲醛转化率为67%,选择性为80%,异丁烯选择性为80%,同比条件下催化剂活性组分流失率减少21%。
以上对本发明提供的一种碳层包覆的复合型催化剂及其制备方法、在催化合成异戊二烯中的应用进行了详细的介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想,包括最佳方式,并且也使得本领域的任何技术人员都能够实践本发明,包括制造和使用任何装置或系统,和实施任何结合的方法。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。本发明专利保护的范围通过权利要求来限定,并可包括本领域技术人员能够想到的其他实施例。如果这些其他实施例具有不是不同于权利要求文字表述的结构要素,或者如果它们包括与权利要求的文字表述无实质差异的等同结构要素,那么这些其他实施例也应包含在权利要求的范围内。
一种碳层包覆的复合型催化剂及其制备方法、在催化合成异戊二烯中的应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0