

IPC分类号 : C12N1/21,C12N15/63,C12P7/62,C12P21/00,C12P9/00,C12P3/00,C12R1/19N,C12R1/38N,C12R1/07N




专利摘要
本发明公开了一种通过增大细菌体积而增加微生物胞内内含物积累量的方法。本发明提供了一种提高微生物胞内内含物积累量的方法,为通过改造影响微生物体积大小的基因增大微生物体积,实现提高微生物胞内内含物积累量。实验证明,本发明构建增大细菌体积的工程菌可以提高聚3羟基丁酸酯(PHB)、蛋白质、聚磷酸和碳小体等的产量,有的工程菌PHB最高可达细胞干重的80%,提高了30%。且本发明的生产工艺简单,成本低廉,应用前景广阔。
权利要求
1.一种提高微生物胞内内含物积累量的方法,为通过改造影响微生物体积大小的基因增大微生物体积,实现提高所述微生物胞内内含物积累量;
所述通过改造影响微生物体积大小的基因增大微生物体积为改变细胞分裂的基因,所述改变细胞分裂的基因的方式包括分别或结合删除和过表达细胞分裂基因、细胞骨架合成基因以及障碍细胞体积增大的细胞壁合成相关基因;
所述方法为敲除出发菌基因组中的
所述出发菌为大肠杆菌。
2.根据权利要求1所述的方法,其特征在于:所述敲除出发菌基因组中的
3.根据权利要求2所述的方法,其特征在于:
所述同源重组中,用于敲除
4.根据权利要求3所述的方法,其特征在于:向所述出发菌中共同导入
所述重组载体包括驱动
5.根据权利要求4所述的方法,其特征在于:
所述含有PHB合成基因的质粒为pBHR68;
所述重组载体为ptK01,其核苷酸序列为序列表中的序列2。
6.根据权利要求1-5中任一所述的方法,其特征在于:所述内含物为聚3羟基丁酸酯、蛋白质、聚磷酸或碳小体。
7.由权利要求1-6中任一所述的方法制备的重组菌。
8.权利要求7所述的重组菌在提高微生物胞内内含物积累量中的应用;
所述微生物为大肠杆菌。
9.根据权利要求8所述的应用,其特征在于:所述内含物为聚3羟基丁酸酯、蛋白质、聚磷酸或碳小体。
说明书
技术领域
本发明涉及生物技术领域,尤其涉及一种通过增大细菌体积而增加微生物胞内内含物积累量的方法,内含物包括聚3羟基丁酸酯(PHB)、蛋白质、聚磷酸和碳小体等。
背景技术
微生物合成的许多内含物有实际应用,比如内含物聚3羟基丁酸酯(PHB)可以作为生物塑料或者医用植入材料、蛋白质内含物可以是有应用价值的酶或者治疗性的多肽、聚磷酸内含物可以用于提取磷元素作为化肥的成份,碳小体内含物有固定二氧化碳的作用等。但是,微生物个体的狭小空间障碍了人们大量地获得这种微生物内含物。
目前已知的微生物细胞分裂基因主要有:ftsZ、ftsA、ftsQ、sulA、minCD。
微生物细胞壁合成相关基因:盘尼西林结合蛋白(PBPs)基因家族mrcA、mrcB、pbpC、dacB、dacA、dacC、pbpG、dacD、ampC、ampH,异戊二烯isoprene合成必需基因yluB,yacM,yabH,yacN,和yqfP,肽聚糖合成和修饰基因csbB,自溶素表达的调控基因lytR。
结合过表达支撑细胞的骨架基因:mreB、mreC、mreD、ftsI、rodZ、mrdA、mrdB、mbl。
对上述基因的单独或结合敲除和过表达都会引起微生物细胞的形态变化,主要是引起细胞体积的变大。
比如,MreB是很多原核生物中起骨架作用的蛋白,敲除后细胞会变成圆形,在大肠杆菌中敲除mreB基因后,大肠杆菌从杆状变成圆形;在此基础上过表达mreB后,部分细胞形态变得不规则,都比野生形的体积大。
以上原理同样适用于其他细胞分裂基因、骨架合成基因以及障碍细胞体积增大的细胞壁合成相关基因的操作引起的微生物细胞体积增大,比如ftsZ系列基因、mreB、mreC、mreD、ftsI、rodZ、mrdA、mrdB、mbl等删除、过表达和各种结合,都能增大细胞体积。
又如,sulA和minCDE是细胞分裂环形成的抑制基因,诱导表达sulA或/和minCDE,使细胞只分裂不分离,细胞体积进一步增大,可以观察到菌体生长从颗粒状变成的纤维状,胞内空间增大不仅提高了内含物的积累量,而且纤维状细胞有利于下游分离细胞和提取内含物。
细胞体积的变大是否能够导致细胞内部可以容纳更多的内含物,成为目前研究的热点。
发明内容
本发明的目的是提供一种提高微生物胞内内含物积累量的方法。
本发明提供的方法,为通过改造影响微生物体积大小的基因增大微生物体积,实现提高所述微生物胞内内含物积累量。
上述方法中,所述通过改造影响微生物体积大小的基因增大微生物体积为如下1)-3)中至少一种:
1)改变细胞分裂的基因,所述改变细胞分裂的基因的方式包括分别或结合删除和过表达细胞分裂基因、细胞骨架合成基因以及障碍细胞体积增大的细胞壁合成相关基因;
2)改变细胞形状和体积的基因,所述改变细胞形状和体积的基因包括支撑细胞的骨架基因mreB、mreC、mreD、ftsI、rodZ、mrdA、mrdB或mbl;
3)改变细胞形状和体积的基因,所述细胞形状和体积的基因包括细胞分裂环形成的抑制基因sulA、FtsZ蛋白抑制因子Min家族蛋白minCDE、细胞分裂环起始形成基因ftsZ、细胞壁合成相关基因ddlb、mrcA、mrcB、pbpC、dacB、dacA、dacC、pbpG、dacD、ampC或ampH。
上述提高微生物胞内内含物积累量的方法可以为敲除出发菌基因组中的mreB基因,且向所述出发菌中共同导入mreB基因和sulA基因,且向所述出发菌中导入含有PHB合成基因的质粒,得到的重组菌为Escherichia coli JM109SG△mreB(ptk01,pBHR68);其出发菌可以为Escherichia coli JM109SG;
所述含有PHB合成基因的质粒具体为pBHR68。
上述方法中,所述敲除出发菌基因组中的mreB基因通过同源重组实现,mreB基因的上游同源臂的核苷酸序列为序列表中序列1自5’末端第1-57位核苷酸;mreB基因的下游同源臂的核苷酸序列为序列表中序列1自5’末端第1361-1417位核苷酸;
上述方法中,所述向出发菌中共同导入mreB基因和sulA基因为通过重组载体向出发菌中共同导入mreB基因和sulA基因;所述重组载体包括驱动mreB基因表达的启动子PmreB、mreB基因、ParaBAD、sulA基因、pSC101复制子和卡那抗性基因。所述重组载体具体为ptK01,其核苷酸序列为序列表中的序列2。
所述同源重组为将含有mreB上游同源臂、FRT、Km、FRT、mreB下游同源臂的DNA片段(序列1)导入出发菌中,实现敲除出发菌基因组中的mreB基因。
上述提高微生物胞内内含物积累量的方法可以为向所述出发菌中导入mreB基因,且导入含有PHB合成基因的质粒;其出发菌可以为Escherichia coli JM109SG;
所述含有PHB合成基因的质粒具体为pBHR68。
上述方法中,向所述出发菌中导入mreB基因的方法为通过重组载体向所述出发菌中导入;所述重组载体包括驱动mreB基因表达的启动子PmreB、mreB基因、复制子和氯霉素抗性基因。所述重组载体具体为pxr01,其核苷酸序列为序列表中的序列3。
所述提高微生物胞内内含物积累量的方法可以为向所述出发菌中导入minCD基因,且导入含有PHB合成基因的质粒;其出发菌可以为盐单胞菌TD08;
所述含有PHB合成基因的质粒具体为pBHR68。
上述方法中,所述向所述出发菌中导入minCD基因的方法为通过重组载体向所述出发菌中导入MinCD基因;所述重组载体为将含有LacI
所述含有LacI
所述提高微生物胞内内含物积累量的方法可以为敲除出发菌基因组中的sigD、lytE和lytD基因,且导入含有PHB合成基因的质粒;其出发菌可以为枯草芽孢杆菌168;
所述含有PHB合成基因的质粒具体为pBHR68。
上述方法中,所述敲除通过同源重组实现;其中,用于敲除出发菌sigD基因的上游同源臂为序列15自5’末端第20-613位核苷酸,下游同源臂为序列15自5’末端第614-1201位核苷酸;
用于敲除出发菌lytE基因的上游同源臂为序列16自5’末端第20-1025位核苷酸,下游同源臂为序列16自5’末端第1851-2765位核苷酸;
用于敲除出发菌lytD基因的上游同源臂为序列17自5’末端第20-1184位核苷酸,下游同源臂为序列17自5’末端第2265-3436位核苷酸。
所述同源重组具体为将含有SigD上下游同源臂的DNA片段(序列15自5’末端第20bp-1201bp)通过重组载体pCU-SigD(序列15)、含有lytE上游同源臂、抗性基因Spe和lytE下游同源臂的DNA片段(序列16自5’末端第20bp-2765bp)通过重组载体pCU-lytE-spe(序列16)和含有lytD上游同源臂、抗性基因Em和lytD下游同源臂的DNA片段(序列17自5’末端第20bp-3436bp)通过重组载体pCU-lytD-Em(序列17)共同导入168中,通过同源重组替换其基因组上的SigD、lytE和lytD基因;
所述提高微生物胞内内含物积累量的方法可以为敲除出发菌基因组中的dacA、ddlB、ampC、ampH和mrcB基因,且导入含有PHB合成基因的质粒;其出发菌可以为Escherichia coli JM109SG。
所述含有PHB合成基因的质粒具体为pBHR68。
上述方法中,所述敲除出发菌基因组中的dacA、ddlB、ampC、ampH和mrcB基因通过同源重组实现,其中,
用于敲除出发菌基因组中的dacA基因的上游同源臂核苷酸序列为序列5,下游同源臂的核苷酸序列为序列6;
用于敲除出发菌基因组中的ddlB基因的上游同源臂核苷酸序列为序列9,下游同源臂的核苷酸序列为序列10;
用于敲除出发菌基因组中的ampC基因的上游同源臂核苷酸序列为序列11,下游同源臂的核苷酸序列为序列12;
用于敲除出发菌基因组中的ampH基因的上游同源臂核苷酸序列为序列13,下游同源臂的核苷酸序列为序列14;
用于敲除出发菌基因组中的mrcB基因的上游同源臂核苷酸序列为序列7,下游同源臂的核苷酸序列为序列8。
上述同源重组为将含有dacA上游同源臂、FRT、Km、FRT、dacA下游同源臂的DNA片段(序列见实施例3的一)、含有ddlB上游同源臂、FRT、Km、FRT、ddlB下游同源臂的DNA片段(序列见实施例3的一)、含有ampC上游同源臂、FRT、Km、FRT、ampC下游同源臂的DNA片段(序列见实施例3的一)、含有ampH上游同源臂、FRT、Km、FRT、ampH下游同源臂的DNA片段(序列见实施例3的一)、含有mrcB上游同源臂、FRT、Km、FRT、mrcB下游同源臂的DNA片段(序列见实施例3的一)均导入Escherichia coli JM109SG中,通过同源重组替换基因组中的dacA、ddlB、ampC、ampH、mrcB基因。
上述方法中,所述内含物为聚3羟基丁酸酯(PHB)、蛋白质、聚磷酸或碳小体。
上述方法中,所述出发菌或微生物为细菌,所述细菌为大肠杆菌、嗜盐菌、假单胞菌或芽孢杆菌。
由上述的方法制备的重组菌也是本发明保护的范围。
上的重组菌在提高微生物胞内内含物积累量中的应用也是本发明保护的范围。
上述应用中,所述内含物为聚3羟基丁酸酯(PHB)、蛋白质、聚磷酸或碳小体。
所述微生物为革兰氏阳性菌或革兰氏阴性菌,所述革兰氏阳性菌具体为芽孢杆菌,所述革兰氏阴性菌具体为大肠杆菌、嗜盐菌或假单胞菌。
本发明通过基因操作,增大了细菌体积,从而增加了微生物胞内内含物包括聚3羟基丁酸酯(PHB)、蛋白质、聚磷酸或碳小体(Carboxysome)等的积累量。本发明所用的微生物可以是革兰氏阴性和阳性菌、包括但不仅限于大肠杆菌、嗜盐菌、假单胞菌、芽孢杆菌等。
本发明提供的增大细菌体积的方法,主要是通过分别或结合删除和过表达细胞分裂基因、细胞骨架合成基因以及障碍细胞体积增大的细胞壁合成相关基因来实现的。比如从出发菌中通过同源重组法敲除细胞分裂基因和/或者障碍细胞体积增大的细胞壁合成相关基因如mreB、mreC、mreD、ftsI、rodZ、mrdA、mrdB、mbl,还可以在此基础上在质粒上补偿表达敲除的基因和诱导表达分裂环形成的抑制基因,然后将质粒转化到敲除株,得到重组菌A;另一方面,也可在出发菌中过表达细胞分裂基因以及障碍细胞体积增大的细胞壁合成相关基因,比如mreB、mreC、mreD、PBP2、PBP3、RodA、RodZ、mrdA、mrdB、mbl,得到重组菌B。由此产生的重组菌在大量积累内含物的情况下体积不断增大,或者从颗粒状细菌成为纤维丝状细菌,两者都能使内含物产量增加。
本发明中的微生物胞内内含物包括聚3羟基丁酸酯(PHB)、蛋白质、聚磷酸或碳小体(Carboxysome)等,既可由野生微生物合成,也可由外源基因表达获得。
本发明用微生物内含物包括聚3羟基丁酸酯聚3羟基丁酸酯(PHB)、蛋白质、聚磷酸和碳小体等进行了实验,发现增大微生物细胞体积确实使聚3羟基丁酸酯聚3羟基丁酸酯(PHB)、蛋白质、聚磷酸和碳小体等胞内内含物的产量得到提高。
所述sulA基因诱导型启动子为阿拉伯糖启动子,序列如序列表中序列2自5’末端第3785-5017位核苷酸,但不限于阿拉伯糖启动子。
所述出发菌为大肠杆菌,具体为Escherichia coli JM109SG。但不限于大肠杆菌(Escherichia coli)。
所述MreB编码基因通过重组载体ptk01导入所述基因组上mreB敲除的中间菌。
所述敲除方法中的同源重组片段氨基酸序列为序列表中序列1。
所述重组载体ptk01为通过Gibson assembly方法得到的载体。
所述重组载体ptk01和pxr01中mreB基因的启动子序列为序列表中序列2自5’末端第1065-1423位核苷酸。
本发明的实验证明,本发明通过增大细菌体积而增加微生物胞内内含物积累量的方法,可以通过分子(基因)操作来实现,比如改变细胞分裂的基因操作,包括分别或结合删除和过表达细胞分裂基因以及障碍细胞体积增大的细胞壁合成相关基因等;本发明构建增大细菌体积的工程菌可以提高PHB、蛋白质、聚磷酸和碳小体等的产量,有的工程菌PHB最高可达细胞干重的80%,提高了30%。且本发明的生产工艺简单,成本低廉,应用前景广阔。
附图说明
图1为大肠杆菌重组菌构建示意图
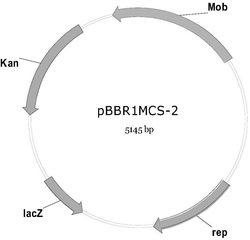
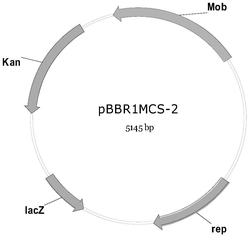
图2为质粒ptk01(左)和PXR01(右)的结果示意图
图3左图为发酵48h后重组菌与对照菌的细胞数目比较,右图为重组菌积累PHB后的沉降速率
图4为扫描电镜观察重组菌积累PHB
图5为透射电镜观察重组菌中PHB的积累方式
图6诱导表达minCD的盐单胞菌TD08菌体体积和PHB积累事宜图
图7为芽孢杆菌变形后形态图
图8为敲除细胞壁合成基因的E.coli JM109SG△dacA△ddlb△AmpC△AmpH△mrcB菌株生产PHB后的形态图
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
图1为大肠杆菌重组菌构建示意图。
所用酶试剂均购自MBI Fermentas公司,提取质粒所用的试剂盒购自北京博迈德科技发展公司,回收DNA片段所用的试剂盒购自美国omega公司,相应的操作步骤按照产品说明书进行;所有培养基如无特别说明均用去离子水配制。
培养基配方:
LB培养基含:5g/L酵母提取物(购自英国OXID公司,产品目录号LP0021),10g/L蛋白胨(购自英国OXID公司,产品目录号LP0042),10g/L NaCl,其余为水。调pH值至7.0-7.2,高压蒸汽灭菌。
MM培养基配置方法:2g/L酵母提取物(购自英国OXID公司,产品目录号LP0021),其余为水。溶解后高压灭菌。冷却后每50ml加入1ml组分I(10g(NH4)2SO4和2gMgSO4加水定容至200ml,高压蒸汽灭菌)和组分II(96.5g Na2HPO4.12H2O和15g KH2PO4,加水定容至200ml,高压蒸汽灭菌)。(NH4)2SO4,MgSO4,Na2HPO4.12H2O,KH2PO4购自国药集团化学试剂有限公司,目录号分别为10002992,10034998,10020392,1017628。
在实际培养过程中,可向上述培养基中再添加一定浓度的抗生素以维持质粒的稳定性,如50μg/mL氨苄青霉素和50μg/mL硫酸卡那霉素。
实施例1、通过改变mreB、sulA和minCD基因增大细胞体积提高内含物产量
一、工程菌的构建
1、敲除出发菌mreB基因构建Escherichia coli JM109SG△mreB
以mreB-Km TF和mreB-KmTR为引物,以pKD13质粒(记载在如下文献中:DatsenkoK&Wanner B(2000)One-step inactivation of chromosomal genes in Escherichiacoli K-12using PCR products.Proceedings of the National Academy of Sciencesof the United States of America97(12):6640-6645.公众可从清华大学获得)为模板,用pfu酶进行PCR反应扩增卡那霉素抗性基因和FRT位点,并在两段设计引入mreB同源臂。
引物为:
mreB-kmTF:
5’
同源臂
CCGGGGATCCGTCGACC3’
mreB-KmTR:
5’
同源臂
TGTAGGCTGGAGCTGCTTCG3’
PCR反应条件:
先95℃预变性5分钟;再95℃变性30秒,58℃退火30秒,72℃延伸1分30秒,30次循环;然后72℃后延伸10分钟。
PCR扩增目的片断(50μL体系)
PCR扩增体系配制时,DNA聚合酶最后加入。
得到1417bp的PCR产物,为含有mreB上游同源臂、Km、FRT、mreB下游同源臂的DNA片段,其核苷酸序列为序列表中序列1,其中,mreB上游同源臂为序列1自5’末端第1-57位核苷酸,Km为序列1自5’末端第487-1281位核苷酸,FRT为序列1自5’末端第86-119和1308-1341位核苷酸,mreB下游同源臂为序列1自5’末端第1361-1417位核苷酸。
将带有重组酶的质粒pKD46recA(Datsenko,K.&Wanner,B.One-stepinactivation of chromosomal genes in Escherichia coli K-12using PCRproducts.Proceedings of the National Academy of Sciences of the United Statesof America97,6640-6645(2000).公众可从清华大学获得)转化到出发菌Escherichiacoli JM109SG(Li,Z.-J.et al.Production of poly(3-hydroxybutyrate-co-4-hydroxybutyrate)from unrelated carbon sources by metabolically engineeredEscherichia coli.Metabolic engineering12,352-359(2010).公众可从清华大学获得)中,得到转化子。
取500μl转化子菌液接种到20ml新鲜LB培养基中,30℃培养至OD6000.6-0.8时加入终浓度为0.2%的阿拉伯糖诱导,诱导后2h,放冰上预冷30分钟,收集3ml菌体,5000rpm离心2分钟,预冷的去离子水洗两次,预冷的10%甘油洗一次,保留100ul甘油,加入10ul含有mreB上游同源臂、Km、mreB下游同源臂FRT的DNA片段,混匀后转移到预冷的电击杯中,电击1.8KV,5ms。加入1ml无抗MMG培养基,37℃培养90分钟,涂MMG卡纳抗性平板,得到转化子。
将转化子进行菌落PCR鉴定,以mreBTF和KmTR为引物,用Taq mix进行PCR反应,得到700bp左右的条带的为阳性克隆。
引物为:
mreBTF:5’CATTAAGCCTTTCTGGACTC3’
KmTR:5’TGACGGCGGTATCCATAT3’
将PCP20质粒(Datsenko,K.&Wanner,B.One-step inactivation of chromosomalgenes in Escherichia coli K-12using PCR products.Proceedings of the NationalAcademy of Sciences of the United States of America97,6640-6645(2000).公众可从清华大学获得.)转入阳性克隆中,通过FRT重组去除基因组上的抗性基因,得到重组菌。
对重组菌基因组上mreB位置进行测序,结果为基因组上删除930bp mreB基因的为阳性重组菌,即为mreB基因敲除的菌株,命名为Escherichia coli JM109SG△mreB,为将序列表中序列1通过同源重组的方式替换JM109SG基因组上mreB基因,实现敲除mreB基因得到的重组菌。
将Escherichia coli JM109SG△mreB扫描电镜观察,结果为mreB基因敲除的大肠杆菌从杆状变成球状,结果如图1所示。
研究发现,在野生型大肠杆菌中过表达mreB,由于原本的细胞骨架的存在,只能在一定程度内使细胞体积变大;基因组上敲除mreB,大肠杆菌从杆状变成圆形,但生长状态不好。在质粒上补偿表达mreB基因,可以改善生长状态,而且由于原本的细胞骨架被破坏,在质粒上补偿表达的MreB蛋白形成的细胞骨架相比野生的要弱,PHB等微生物内容物的积累容易使细胞骨架发生形变,从而降低PHB颗粒之间的空间位阻,PHB颗粒增大增多。在发酵过程中,本发明确实观察到了mreB补偿表达的重组菌可由于PHB积累而撑起变成圆球形,具体实验如下。
2、表达载体ptk01、ptr01和pSEVA341-LacI
1)、表达载体ptk01的制备
(1)目的基因mreB的获得
提取大肠杆菌K-12系JM109SG(Escherichia coli JM109SG)的基因组为模板,以G-mreB-1TF和G-mreB-2TR为引物,用pfu酶进行PCR扩增。
引物为:
G-mreB-1TF:5’tcctattccgaagttcctatcttcccctgcctgcatccg3’
G-mreB-2TR:5’gtcaattcagggtggtgaatCTCACAGCCACTTGATACTAACGTG3’
得到1443bp的PCR产物,为目的基因mreB和驱动其表达的启动子PmreB,目的基因mreB的核苷酸序列为序列表中序列2自5’末端第21-1064位,启动子PmreB的核苷酸序列为序列表中序列2自5’末端第1065-1423位核苷酸。
(2)目的基因sulA的扩增
以Escherichia coli JM109SG基因组为模板,以G-sulA-1TF和G-sulA-1TR为引物,用pfu酶进行PCR扩增目的基因sulA。
G-sulA-1TF:5’ctctaaggaggttataaaaaatgTACACTTCAGGCTATGCACATC3’
G-sulA-1TR:5’ttcaaaagcgctctgaagttGCGGAAACCGCTTCAGACAAG3’
得到610bp的PCR产物,为目的基因sulA基因,其核苷酸序列为序列表中序列2自5’末端第5018-5527位。
(3)pSC101复制子和ParaBAD的扩增
以G-ptk ori-2TF和G-ptk ori-1TR为引物,用ptkRED为模板,用Q5超薄真2XMaster Mix扩增质粒复制子。
G-ptk ori-2TF:5’TAGTATCAAGTGGCTGTGAGattcaccaccctgaattgactctc3’
G-ptk ori-1TR:5’GCATAGCCTGAAGTGTAcattttttataacctccttagagctcgaattcc3’
得到3594bp的PCR产物,其核苷酸序列为序列表中序列2自5’末端第1424-5017位,pSC101复制子核苷酸序列为序列表中序列2自5’末端第1609-3303位和ParaBAD核苷酸序列为序列表中序列2自5’末端第3785-5017位。
(4)抗性基因Km的扩增
以G-Km-1TF和G-Km-1TR为引物,以pKD13质粒为模板,用pfu酶进行PCR扩增抗性基因Km。
G-Km-1TF:5’ttgtctgaagcggtttccgcaacttcagagcgcttttgaa3’
G-Km-1TR:5’tcggatgcaggcaggggaagATAGGAACTTCGGAATAGGA3’
得到1205bp的PCR产物,抗性基因Km为序列表中序列2自5’末端第6001-6795位。
将上述(1)、(2)、(3)、(4)的DNA片段通过Gibson assembly同源重组方法连接,得到连接产物,转化到大肠杆菌Trans1-T1中,得到转化子。
提取转化子的质粒,送去测序,结果为获得含有上述4个片段,该质粒记作ptk01(又称为ptk-mreB-ara-sulA),其核苷酸序列为序列表中的序列2,质粒的结构示意图见图2的左图。
2)、表达载体pxr01的制备
(1)mreB和启动子PmreB
提取大肠杆菌K-12系JM109SG(Escherichia coli JM109SG)(本实验室保存)的基因组,以Escherichia coli JM109SG基因组为模板,以p15a-mreB F和p15a-mreB R为引物,用pfu酶进行PCR扩增。
p15a-mreB F:5’ttagaactgcgattcttcagctcacagccacttgatactaacgtg3’
p15a-mreB R:5’tattggtgcccttaaacgcccttcccctgcctgcatcc3’
得到1443bp的PCR产物,为目的基因mreB和驱动其表达的启动子PmreB,具有序列表中序列3自5’末端第2126-3548位核苷酸,目的基因mreB的核苷酸序列为序列表中序列3自5’末端第2485-3528位,启动子PmreB的核苷酸序列为序列表中序列3自5’末端第2126-2484位核苷酸。
(2)含有复制子和氯霉素抗性基因的目的片段的获得
以mreB-p15a F和mreB-p15a R为引物,以p15AMBA-aceA质粒为模板,用pfu酶进行PCR扩增,得到2125bp的含有复制子和氯霉素抗性基因的目的片段,其中,复制子的核苷酸序列为序列表中序列3自5’末端第1324-1868位,氯霉素抗性基因的核苷酸序列为序列表中序列3自5’末端第36-695位核苷酸。
引物为:
mreB-p15a F:5’tcggatgcaggcaggggaagggcgtttaagggcaccaataac3’
mreB-p15a R:5’tagtatcaagtggctgtgagctgaagaatcgcagttctaaaagc3’
将上述(1)和(2)的两个片段通过Gibson assembly同源重组方法连接,得到连接产物,转化到大肠杆菌Trans1-T1中,得到转化子。
提取转化子的质粒,送去测序,结果为获得含有上述2个片段的质粒,记作pxr01,其核苷酸序列为序列表中序列3,该质粒的结果示意图见图2的右图。
3)、表达载体pSEVA341-LacI
(1)minCD基因的扩增
采用在盐单胞菌HalomonasTD8中建立的IPTG诱导的LacI
以TD8野生型基因组为模板,MinCD-F和MinCD-R为引物进行PCR扩增,得到1752bp的PCR产物,即为minCD基因,其核苷酸序列为序列表中序列4自5’末端第3189-4914位核苷酸;
以pSEVA434为模板,trcF和trcR为引物进行PCR扩增,得到1458bp的PCR产物,即为LacI
以minCD基因和LacI
用XbaI和SacI酶切LacI
将阳性转化子进行菌落PCR鉴定,引物R24,F24进行菌落P CR验证,得到3301bp的为阳性克隆。将阳性克隆提取质粒送去测序,该质粒为将序列表中序列4所示的核苷酸LacI
表1为克隆minCD的扩增引物序列
下划线表示SOE PCR的互补重叠区,也是26bp的SD序列。加粗表示酶切位点。
3、工程菌Escherichia coli JM109SG△mreB(ptk01,pBHR68)、E.coli JM109SG(pxr01,pBHR68)和TD08(pSEVA341-LacI
pBHR68载体记载在如下文献中:Spiekermann P,Rehm BHA,Kalscheuer R,Baumeister D,Steinbüchel A(1999)A sensitive,viable-colony staining methodusing Nile red for direct screening of bacteria that accumulatepolyhydroxyalkanoic acids and other storage com-pounds.Arch Microbiol171:73-80,公众可从清华大学获得,该质粒中含有来源于罗氏真养杆菌Ralstoniaeutropha的PHB合成基因phaC2基因、β-酮基硫解酶PhaA和NADPH依赖的乙酰乙酰辅酶A还原酶phaB。
将上述2制备的表达载体ptk01和pBHR68用电击转化法同时转入到上述1获得的mreB敲除的重组菌Escherichia coli JM109SG△mreB中,获得重组菌A,为敲除mreB且质粒过表达mreB和sulA基因;
将上述2获得的表达载体ptk01和pBHR68用电击转化法同时转入到Escherichiacoli JM109SG中,得到对照菌E.coliJM109SG(ptk01,pBHR68),质粒过表达mreB和sulA基因;
将上述2获得的表达载体pxr01和pBHR68用电击转化法同时转入到E.coliJM109SG中,获得重组菌B,质粒过表达mreB基因。
将步骤二获得的pSEVA341-LacI
将pSEVA341接合转化入盐单胞菌TD08,得到对照菌株TD08(pSEVA341)。
分别将重组菌A、B涂布于LB-Amp,Km的固体培养平板上,重组菌A在30℃培养12小时,重组菌B在37℃培养12小时;分别挑取在LB-Amp,Km固体培养平板上长出的单克隆,将其接种到LB-Amp,Km液体培养基中,重组菌A在30℃、200rpm下摇床培养12小时,重组菌B在37℃、200rpm下摇床培养12小时。
重组菌A提取质粒用BglII单酶切,得到6.8kb和8.1kb的片段,证明ptk01和pBHR68已经被成功转入到mreB敲除的重组菌Escherichia coli JM109SG△mreB中,命名为:Escherichia coli JM109SG△mreB(ptk01,pBHR68)(图1),其为敲除Escherichia coliJM109SG基因组的mreB基因,且通过质粒ptk01将mreB基因和sulA基因与质粒pBHR68共同导入E.coli JM109SG中得到的重组菌;
重组菌B提取质粒用NdeI单酶切,得到3.5kb、8.1kb的片段,证明pxr01和pBHR68已经被成功转入到E.coliJM109SG中,命名为Escherichia coli JM109SG(pxr01,pBHR68),其为将mreB基因通过质粒pxr01与质粒pBHR68共同导入E.coli JM109SG中得到的重组菌。
将pBHR68转入E.coliJM109SG得到Escherichia coli JM109SG(pBHR68),提取质粒并用BglII酶切验证,得到8.1kb的片段,证明为阳性。
二、工程菌Escherichia coli JM109SG△mreB(ptk01,pBHR68)、E.coli JM109SG(pxr01,pBHR68)和TD08(pSEVA341-LacI
A、工程菌Escherichia coli JM109SG△mreB(ptk01,pBHR68)、E.coli JM109SG(pxr01,pBHR68)提高内含物产量
1、LB培养基条件下摇瓶培养PHB产量检测
1)PHB产量检测
表达工程菌E.coliJM109SG(pBHR68)、E.coli JM109SG(ptk01,pBHR68)、E.coliJM109SG△mreB(ptk01,pBHR68)分别用LB培养基,在30℃,200rpm过夜培养;然后按5%接种量(v/v),分别接种至50mL的含葡萄糖的LB培养基(含:5g/L酵母提取物,10g/L蛋白胨,10g/L NaCl,20g/L葡萄糖,其余为水,pH7.0-7.2),30℃,200rpm,摇瓶培养48小时,得到菌液,经离心、水洗、冰干后得到干细胞待测样品,每个菌种设置三组平行。运用气相色谱仪(GC,Hewlett-Packard model6890)验证细胞中均聚物的积累。运用液相色谱仪(HPLC,SpectroSYSTEM,Thermo Separation Products,USA)检测葡萄糖的消耗。
液相检测色谱柱为Bio-rad公司有机酸柱,Aninex HPX-87X Ion ExclusionColumn(300×7.8mm)。流动相为5mM H2SO4,流速为0.5ml/min。进样量为10μl。检测器使用美国SPECTRA SYSTEM公司的RI-150示差检测器。
样品检测:取1.5ml菌液,10000rpm离心后取上清液,用0.45mm滤膜过滤后作为色谱的样品。
标准曲线测定:葡萄糖水溶液。将葡萄糖的标准样品稀释不同倍数,以0.5ml/min的流速,通过HPLC检测,并绘制标准曲线。
葡萄糖标样的出峰时间为第11.051min。
结果为各组葡萄糖几乎全部用完,根据标样定量检测出各组培养基中葡萄糖的浓度,用于监测培养基中碳源的变化从而适当的补加碳源。
气相检测3-羟基丁酸均聚物的方法:
设定炉温为80℃,进样器温度为200℃,检测器温度为220℃,柱头压力为0.25Mpa,程序升温条件为:80℃停留1.5分钟,以30℃/min的速度升温至140℃,接着以40℃/min的速度升温至220℃并在此温度保持0.5分钟。样品的进样量为1μl,使用安捷伦公司生产的微量进样器。
气相样品准备:取40-60mg待测样品的干细胞(菌液10000rpm,10分钟条件下离心,所得细胞沉淀水洗一次之后,冰干,得到干细胞,均聚物产于细胞中),加2ml氯仿,2ml酯化液(纯甲醇中含3%(v/v)的浓硫酸及2g/L苯甲酸作内标)于酯化管中,加盖密封后于100下加热4小时。冷却后加入1ml蒸馏水,充分振荡后静置,待氯仿相与水相完全分层后,取下层氯仿相1μl注入气相色谱仪(HP公司Hewlett Packard6890)中进行色谱分析。依照HP公司Hewlett Packard6890气相色谱仪的说明书操作气相色谱仪。
标准样品准备:取10-20mg的3-羟基丁酸3HB(sigma-aldrich,产品货号:363502-10G)水溶液于酯化管中,加2ml氯仿,2ml酯化液,加盖密封后在100℃进行酯化。在本实施例中的气相检测条件下,标准样品在第2.2分钟有明显出峰,表征酯化样品中的3-羟基丁酸。
以标准样品为对照,如果待测细胞的酯化样品(待测样品)在2.2分钟也有明显的出峰,且无其它特异峰出现,说明待测细胞的酯化样品有且仅有3HB单体。因为在干细胞中3HB只能以聚合物的形式存在,这样的结果即证明了均聚物的产生。
结果为E.coli JM109SG△mreB(ptk01,pBHR68)、E.coli JM109SG(ptk01,pBHR68)、E.coli JM109SG(pBHR68)的干细胞在2.2分钟也有明显的出峰,并且没有其它特异出峰,说明能够生产均聚物。
通过分析气相检测到的出峰面积和样品量,可得到干细胞中含有均聚物的比重(wt%),及每L培养体系中能够生产的均聚物或共聚物的浓度(g/L),具体结果见表1所示。
在LB培养基中培养E.coli JM109SG(pBHR68)、E.coli JM109SG(ptk01,pBHR68)、E.coliJM109SG△mreB(ptk01,pBHR68)菌株,4h后E.coli JM109SG(ptk01,pBHR68)和E.coli JM109SG△mreB(ptk01,pBHR68)中加入0.2%的阿拉伯糖诱导表达sulA基因,细胞开始变长,10h后加入终浓度为20g/L的Glucose,48h后将菌液离心、水洗、冰干后得到干细胞待测样品,按照上述“气相样品准备”得到待测样品,以标准样品的气相检测结果为参照,分析这些菌株中均聚物的积累量。
结果发现敲除基因组上mreB后又在质粒上补偿表达mreB的菌株E.coli JM109SG△mreB(ptk01,pBHR68)的细胞干重CDW和3HB产量都比对照菌E.coli JM109SG(pBHR68)和E.coli JM109SG(ptk01,pBHR68)有明显提高,得到8g/L3-羟基丁酸(3HB),结果见表2。
在含有20g/L葡萄糖的LB培养基中培养E.coliJM109SG(pBHR68)、E.coli JM109SG(pxr01,pBHR68)菌株,48h后将菌液离心、水洗、冰干后得到干细胞待测样品,按照上述“气相样品准备”得到待测样品,以标准样品的气相检测结果为参照,分析这些菌株中均聚物的积累量。
结果发现E.coli JM109SG(pxr01,pBHR68)的3HB百分含量比对照菌E.coliJM109SG(pBHR68)有明显提高,得到7g/L3-羟基丁酸(3HB),结果也如表2所示。
表2为LB培养基条件下摇瓶培养的检测结果
由表2的测定结果可以看出,大肠杆菌E.coli JM109SG(pxr01,pBHR68)单纯过表达mreB基因,细胞3HB百分含量比对照菌E.coli JM109SG(pBHR68)有明显提高;而在基因组上敲除mreB又在质粒上补偿表达mreB基因的E.coli JM109SG△mreB(ptk01,pBHR68)的细胞干重和PHB百分含量都有更显著的提高。
2、重组菌积累PHB后的形态检测
电镜样品准备:E.coli JM109SG(pBHR68)、E.coli JM109SG(ptk01,pBHR68)、E.coli JM109SG△mreB(ptk01,pBHR68)发酵48小时后收集100ul菌体,5000rpm,离心2分钟,用戊二醛固定2小时;PBS洗两次,每次10分钟;梯度乙醇脱水:50%、70%、80%、90%每个梯度3-5分钟,100%乙醇脱水3次,每次3-5分钟;叔丁醇:乙醇=1:1处理10分钟;纯叔丁醇处理10分钟;最后用叔丁醇覆盖菌体,放-20℃冷冻20分钟以上,然后冷冻干燥成粉末状。得到冰干的菌体。
将上述菌体用牙签沾取合适的量粘在导电胶上,表面喷金60秒,进行扫描电镜观察。
同一放大倍数的结果如图4所示,WT:E.coli JM109SG(pBHR68);JM109SG+mreB+sulA:E.coli JM109SG(ptk01,pBHR68);JM109SG△mreB+mreB+sulA:E.coli JM109SG△mreB(ptk01,pBHR68),可以看出,与W T比,重组菌E.coli JM109SG△mreB(ptk01,pBHR68)积累聚3-羟基丁酸PHB后体积明显变大,有些细菌由于积累PHB,形态从杆状被撑成圆形。
3、透射电镜观察重组菌中PHB的积累方式
E.coli JM109SG(pBHR68)、E.coli JM109SG(ptk01,pBHR68)、E.coli JM109SG△mreB(ptk01,pBHR68)发酵48小时后收集500微升菌体,5000rpm,离心2min,用戊二醛固定,然后用树脂包埋,做超薄切片,进行透射电镜观察。
结果如图5所示,WT:E.coli JM109SG(pBHR68);JM109SG△mreB+mreB+sulA:E.coli JM109SG△mreB(ptk01,pBHR68),可以看出,与W T比,重组菌E.coli JM109SG△mreB(ptk01,pBHR68)中被3-羟基丁酸PHB撑成圆形的大肠杆菌中充满3-羟基丁酸PHB颗粒,并且平均体积比对照菌大。
4、MM培养基条件下摇瓶培养PHB产量检测
1)PHB产量
表达工程菌E.coli JM109SG(pBHR68)、E.coli JM109SG(ptk01,pBHR68)、E.coliJM109SG△mreB(ptk01,pBHR68)分别用LB培养基,在30℃,200rpm过夜培养;然后按5%接种量(v/v),分别接种至50mL的含葡萄糖的MM培养基(含:2g/L酵母提取物,1ml组分I,1ml组分II,20g/L葡萄糖,其余为水),30℃,200rpm,5小时后E.coli JM109SG(ptk01,pBHR68)和E.coli JM109SG△mreB(ptk01,pBHR68)中加入0.2%的阿拉伯糖诱导表达sulA基因,细胞开始变长,10小时后加入终浓度为20g/L的Glucose,48小时后将菌液离心、水洗、冰干后得到干细胞待测样品,并通过气相色谱检测(条件和方法同1),结果如表3所示。每个菌种设置三组平行。
可以看出,通过增大细菌体积来提高内含物PHB产量。
表3为MM培养基条件下摇瓶培养的检测结果
2)、细胞数目和沉降速度检测
将发酵48h后的E.coli JM109SG△mreB(ptk01,pBHR68)涂LB平板过夜培养检测细胞数目和静置30分钟观察沉降速率。以E.coli JM109SG(pBHR68)为对照。
检测细胞数目结果如图3左图所示,E.coli JM109SG△mreB(ptk01,pBHR68)仅是E.coli JM109SG(pBHR68)的1/3,为7X10
观察沉降速率结果如图3右图所示,E.coli JM109SG△mreB(ptk01,pBHR68)沉降速率比对照菌E.coli JM109SG(pBHR68)大。
从上述结果可以看出,E.coli JM109SG△mreB(ptk01,pBHR68)细胞个数较少,但细胞干重和PHB含量均有明显提高,说明每个细胞的PHB的积累增加;且其可以维持诱导后的长度,沉降速率比对照菌E.coli JM109SG(pBHR68)大,这就为下游的分离提取提供便利。
由表3的结果可以看出,MM培养条件下,基因组上敲除mreB又在质粒上补偿表达mreB基因的E.coli JM109SG△mreB(ptk01,pBHR68)的细胞干重和PHB百分含量都有显著的提高,而且细胞数目仅是对照菌的1/3;而且在基因组上敲除mreB又在质粒上补偿表达mreB基因同时诱导表达sulA基因的E.coli JM109SG△mreB(ptk01,pBHR68)可以维持长度,所以沉降速率比对照菌E.coli JM109SG(pBHR68)大,这就为下游的分离提取提供便利。理论上,180g葡萄糖能转化成86g3-羟基丁酸,理论上的碳源转化率为86/180X100%=48%。由表3可知E.coli JM109SG△mreB(ptk01,pBHR68)的碳源转化率为8.11/20X100%=40.5%。
5、E.coli JM109SG△mreB(ptk01,pBHR68)生产聚磷酸、碳小体(Carboxysome)等内含物
聚磷酸(Polyphosphate,Polyp)是一种线性的聚合分子,由多个磷酸基团通过高能磷酸键连接而成,普遍存在于生物体内,聚磷酸能够达到细胞干重的15%。聚磷酸的合成酶有聚磷酸激酶(PolyphosphateKinase,PPK);外切聚磷酸酶(Exopolyphosphase,PPX)、内切聚磷酸酶(Endopolyphosphase,PPN)。
在生物污泥中利用过表达聚磷酸激酶工程菌富集大量聚磷酸,有利于污水处理和磷酸环境修复,从而避免了利用金属离子处理带来的负面污染,在重组菌A中过表达聚磷酸激酶基因,聚磷酸的积累量占细胞干重的百分比可由原来的15%提高到40%。
根据文献报道,carboxysome是存在于一些自养细菌细胞内的多角形或六角形内含体,该结构是自养细菌所特有的内膜结构,大小约100nm。carboxysome由以蛋白质为主的单层膜包围,内含固定二氧化碳所需的1,5-二磷酸核酮糖羧化酶和5-磷酸核酮糖激酶,是自养型细菌固定二氧化碳的部位。因而,carboxysome可以固定二氧化碳,提高碳源的利用率。
在PHB的生产中,大量的碳源在大肠杆菌中转化为其他代谢产物,并没有转化为PHB,碳源的利用率很低是导致PHB生产成本较高的原因之一。为了扩大PHB的生产,提高碳源的转化率具有重大的意义。
在生产PHB的大肠杆菌中构建carboxysome,从而将二氧化碳在细菌体内进行固定,再次利用。大肠杆菌本身是不能合成carboxysome的,其并不能固定二氧化碳,通过调研,合成carboxysome相关的基因共有10个(cbbL、cbbS、csoS2、csoS3、csoS4A、csoS4B、csoS1C、csoS1A、csoS1B、csoS1D),而将这10个基因构建在质粒上,转入大肠杆菌内形成碳小体。
将含有这10个基因的质粒转化到E.coli JM109SG△mreB(ptk01,pBHR68)和对照菌E.coli JM109SG(pBHR68),分别得到含有合成carboxysome相关的基因的E.coliJM109SG△mreB(ptk01,pBHR68)和含有合成carboxysome相关的基因的E.coli JM109SG(pBHR68),在含有20g/L的LB培养基中发酵48h后,检测PHB的积累量,方法同上。
结果如下:
含有合成carboxysome相关的基因的E.coli JM109SG△mreB(ptk01,pBHR68)的碳源转化率为45%;
含有合成carboxysome相关的基因的E.coli JM109SG(pBHR68)的碳源转化率为25%。
因此,E.coli JM109SG△mreB(ptk01,pBHR68)的碳小体的积累量有较明显提高,因为碳小体增多可以提高碳源转化率。
B、工程菌盐单胞菌TD08(pSEVA341-LacI
1、TD08(pSEVA341-LacI
表达工程菌盐单胞菌TD08(pSEVA341-LacI
表4过表达minCD的重组菌生产PHB的摇瓶实验结果
由上表可知,加入IPTG诱导,过表达minCD的重组TD08菌株的细胞干重和只含有空载体的TD08菌株没有明显差别,表明相比于生长早期加入诱导剂,稳定期后加入并不会影响细胞的正常生长;同时,过表达MinCD的重组菌株的PHB含量明显高于只含有空载体的普通TD08菌株,基本维持在80wt%左右,而只含有空载体的普通TD08只有69wt%。加入诱导剂组PHB含量为82wt%,与空载体有显著性差异(P value<0.05)。没加入诱导剂组的TD08(pSEVA341-MinCD)由于存在本地表达,即使不诱导产量也得到提高。
诱导表达minCD的实验组的菌体体积明显增大,PHB积累量增加,结果如图6所示。
2、TD08(pSEVA341-LacI
表面按活性剂被誉为工业味精,具有固定的亲水端和疏水端,在溶液的表面能定向排列并显著减低溶液的表面张力。表面活性剂具有增粘性增泡性抗硬水性消毒杀菌助悬润湿曾柔以及去垢洗涤等作用,是一类被广泛使用的化工用品。表面按活性剂被誉为工业味精,具有固定的亲水端和疏水端,在溶液的表面能定向排列并显著减低溶液的表面张力。表面活性剂具有增粘性增泡性抗硬水性消毒杀菌助悬润湿曾柔以及去垢洗涤等作用,是一类被广泛使用的化工用品。
phaR可溶性蛋白具有较强的乳化作用,phaR包涵体、含有phaR的细胞裂解液也具有较强的乳化作用。phaR能够在95℃下保持较好的乳化功能,且结构稳定性良好。
将TD08(pSEVA341-LacI
将发酵产物菌体离心收集,并超声破碎,phaR是一种可溶性蛋白,离心后分别取上清进行乳化实验,TD08(pSEVA341-LacI
可以看出,TD08(pSEVA341-LacI
实施例2、增大枯草芽孢杆菌的体积提高PHB内含物的含量
一、重组菌枯草芽孢杆菌168△SigD△lytE△lytD(pBHR68)的构建
1、168△SigD的构建
以枯草芽孢杆菌168(Anagnostopoulos and J.Spizizen.1961.Requirement fortransformation in Bacillus subtilis.J.Bacteriol.81:741-746,公众可从清华大学获得)为模板,分别用如下引物对进行PCR扩增:
SigD-up-F:gcatgcctgcaggtcgactagctgaaagcgcatatgttta
SigD-up-R:AGCAGATTCTTTAATTTTCCCCCTAATACCTTAATTA
SigD-down-F:ggtattagggggaaaattaaagaatctgctggaaaaag
SigD-down-R:CGAATTCGAGCTCGGTACCCAACACAGCTTTATCCGACA
分别得到594bp的SigD上游同源臂(序列15自5’末端第20bp-613bp位核苷酸)、588bp的SigD下游同源臂(序列15自5’末端第614bp-1201bp位核苷酸)、另外使用限制性内切酶xbaI和smaI对质粒载体pCU进行双酶切,将环形质粒pCU变为4.2kb的线性片段pCU(xbaI smaI)。
将这三个PCR片段通过试剂盒Gibson master mix(NEB)重组连接,得到质粒pCU-SigD,经过测序,该质粒的核苷酸序列为序列表中序列序列15,其为将含有SigD上下游同源臂的DNA片段(序列15自5’末端第20bp-1201bp)插入pCU载体的xbaI和smaI位点得到的质粒。
将质粒pCU-SigD转入枯草芽孢杆菌168中,该质粒与168基因组进行同源重组,得到重组菌,经过测序,该重组菌缺失SigD基因,命名为168△SigD。
2、168△SigD△lytE△的构建
以枯草芽孢杆菌168为模板,分别用如下引物对进行PCR扩增:
lytE-up-F:gcatgcctgcaggtcgactgtggttaatttcagacgtggc
lytE-up-R:AATTTTATTGCAATAATTTTCCTCCCCAAATGTTAACTC
Spe-F:tattgcaataaaattagcctaattg
Spe-R:GAACATAATCAACGAGGTGAAA
lytE-down-F:tcacctcgttgattatgttctttttagagaaaacccgttc
lytE-down-R:CGAATTCGAGCTCGGTACCCATGGCTTCACACCTTTGTGAC
分别得到1006bp的lytE上游同源臂(为序列16自5’末端第20bp-1025bp位核苷酸)、915bp的lytE下游同源臂(为序列16自5’末端第1851bp-2765bp位核苷酸)、825bp的抗性基因Spe(为序列16自5’末端第1026bp-1850bp位核苷酸)。
将这三个PCR片段和酶切后的载体片段pCU(xbaI smaI)通过试剂盒Gibsonmaster mi
一种通过增大细菌体积而增加微生物胞内内含物积累量的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0