








专利摘要
本发明涉及适用于递送分子至哺乳动物的组合物和方法。本发明具体地涉及肽衍生物(肽和伪肽)、其二聚体或多聚体以及它们作为感兴趣分子的载体的用途。本发明还涉及缀合物,其含有连接至感兴趣分子的本发明的肽衍生物或其二聚体或多聚体。本发明的肽具体地可通常以前药缀合物的形式用于经特异性受体(受体介导的转运,RMT)将感兴趣的药物或诊断分子例如治疗分子、成像或诊断剂或分子探针负载穿过健康或病理(癌细胞)的不同组织或器官(例如肝、肾上腺和肠)的细胞膜,且具体地使其穿过神经系统的生理屏障例如血脑屏障(BBB)、血-脊髓屏障(BSCB)或血-视网膜屏障(BRB)。
权利要求
1.肽或伪肽,其包含选自SEQIDNO:1-5中任一个的序列。
2.具有下式(A)的多聚体试剂:
Ph-M-Pi(A)
其中,
各P表示相同或不同的肽或伪肽,其包含氨基酸序列:A1-Met-A2-Arg-Leu-Arg-A3-A4,其中A1和A4独立地表示半胱氨酸或其类似物或其电子等排体,A2表示脯氨酸或其类似物或其电子等排体,且A3表示甘氨酸或其类似物或其电子等排体;
M是分子平台,和
h和i是彼此独立地选自1、2、3、4或5的整数。
3.权利要求2的多聚体试剂,其中A1和A4独立地表示Cys或其类似物,其选自:(D)-cys、青霉胺(Pen)和(D)-青霉胺((D)-Pen),和/或其中A2表示Pro类似物,其选自:哌可酸(Pip)和噻唑烷-4-羧酸(Thz);和/或其中A3表示Gly或肌氨酸(Sar)。
4.权利要求2或3的多聚体试剂,其中h和i彼此独立地为1或2,优选地,h=i=1。
5.权利要求2-4中任一项的多聚体试剂,其中P为具有以下通式(I′)的肽:
(D)-Cys-Met-A2-Arg-Leu-Arg-A3-A4(I′)
其中A2表示Pro、Pip或Thz,A3表示Gly或Sar且A4表示Cys、(D)-Cys、Pen或(D)-Pen。
6.权利要求2-5中任一项的多聚体试剂,其中P为肽,其包含选自SEQIDNO:1-5中任一个的序列。
7.权利要求2-6中任一项的多聚体试剂,其中M是多官能有机化合物,其具有至少2个活性官能团,优选至少3个活性官能团。
8.权利要求2-7中任一项的多聚体试剂,其中M为聚赖氨酸或三(2-氨基乙基)氨基。
9.权利要求2-8中任一项的多聚体试剂,其为同源二聚体。
10.前述权利要求中任一项的肽、伪肽或多聚体试剂,其中所述肽或伪肽含有至少一个拟肽键,选自:亚甲基(-CH2-)或磷酸酯(-PO2-)、仲胺(-NH-)或氧基(-O-)、α-氮杂肽、α-烷基肽、N-烷基肽、膦酰胺酯、缩肽、羟基亚甲基、羟基乙烯、二羟基乙烯、羟基乙胺、逆反肽、亚甲氧基、酮亚甲基、酯、次膦酸酯、次膦酸、膦酰胺或卡巴类似物。
11.前述权利要求中任一项的肽、伪肽或多聚体试剂,其中所述肽或伪肽的N端(N末端)官能团经酰化保护和/或所述肽或伪肽的的C-端(C-末端)官能团经酰胺化或酯化保护。
12.下式(III)的接合化合物:
VxSzDy(III)
其中V表示前述权利要求中任一项的肽或伪肽或多聚体试剂,S表示间隔基,D表示活性物质或感兴趣的物质,x是1至5的整数和y和z是1至10的整数。
12.权利要求11的接合化合物,其特征在于x=z=y=1;x=z>y,y=z>x或z>x>y;或其中z等于0且x和y等于1或y大于x。
13.权利要求11的接合化合物,其特征在于所述活性物质或感兴趣的物质是感兴趣的治疗分子、诊断或医学成像剂、分子探针、化学小分子、肽或多肽、蛋白、抗原、抗体或抗体的一部分、核酸或寡核苷酸、siRNA、miRNA、核酶、标记物或示踪物。
14.权利要求11的接合化合物,其特征在于在V和D之间的偶联,或者一端是V和S而另一端是S和D之间的偶联是通过共价键、离子键、氢键、疏水键或范德华键中的一种或多种实现的,其在生理介质或细胞中发生裂解或不发生裂解。
15.权利要求11的接合化合物,其中D与V偶联,如果需要经由S,在V的N末端和/或C末端中的一个或两者偶联,和/或在构成V的天然或非天然氨基酸侧链所带有的一或多个活性基团偶联。
16.制备接合化合物的方法,其特征在于其包括以下步骤:将权利要求1-10中任一项的肽或伪肽或多聚体与物质D偶联,如果需要经由间隔基S偶联,该偶联通过化学、生物化学或酶途径或通过基因工程进行。
17.药物组合物,其包含至少一种权利要求11的接合化合物和一种或多种药学上可接受的赋形剂。
18.诊断性组合物,其包含由权利要求11的接合化合物所组成的诊断或医学成像剂。
19.递送分子至受试者的方法,其包括向所述受试者给药偶联至权利要求1-10中任一项的肽或伪肽或多聚体的分子。
说明书
本发明涉及适用于递送分子至哺乳动物的组合物和方法。本发明具体地涉及肽衍生物(肽和伪肽)、其二聚体或多聚体以及它们作为感兴趣分子的载体的用途。本发明还涉及缀合物,其含有连接至感兴趣分子的本发明的肽衍生物或其二聚体或多聚体。本发明的肽具体地可通常以前药缀合物的形式用于经特异性受体(受体介导的转运,RMT)将感兴趣的药物或诊断分子例如治疗分子、成像或诊断剂或分子探针负载穿过健康或病理(癌细胞)的不同组织或器官(例如肝、肾上腺和肠)的细胞膜,且具体地使其穿过神经系统的生理屏障例如血脑屏障(BBB)、血-脊髓屏障(BSCB)或血-视网膜屏障(BRB)。
发明背景
根据IMSHealth,在2007年,治疗中枢神经系统(CNS,脑和脊髓)病症的药物的全球市场大约700亿美元,该总额中有近90亿美元代表的是由药物递送技术研发的产品(Jain,2008,JainPharmaBiotechReport,DrugDeliveryinCNSdisorders)。因此,神经病学连同心血管医学和肿瘤学是当下三大治疗领域。尽管全世界患CNS障碍和病症的人数高于患心血管疾病或癌症的人数,神经病学仍然是有待开发的市场。其由以下事实进行部分解释:98%的治疗CNS病症的潜在药物并未穿过血脑屏障或BBB(Pardridge,2003,Mol.Interv.,3,90-105)。目前市场上仅有5%的药物针对神经系统。
事实上,CNS是由于两个主要的生理屏障系统(BBB/BSCB和血-脑脊液屏障(BCSFB))的存在而免受潜在的有毒物质的侵害。所述BBB被认为是在脑水平摄取等离子体配体的主要途径。其表面积约是BCSFB表面积的5000倍。BBB的结构性血管(constitutivebloodvessel)的总长大约600km。大脑皮层每立方厘米所含的相当于1km血管所含。BBB的总表面积估计有20m2(DeBoer等人,2007,Clin.Pharmacokinet.,46(7),553-576)。BBB和BSCB被视为在研发治疗CNS病症和损伤的新型疗法中需要克服的主要障碍(Neuwelt等人,2008,LancetNeurol.,7,84-96)。
类似地,在眼中,血-视网膜屏障(BRB)是血眼屏障的一部分,所述血眼屏障是由紧密结合在一起的细胞所组成并抵御一些物质进入视网膜。所述BRB有两种成分:视网膜血管内皮和视网膜色素上皮,其还被称为内部和外部组分(内部BRB[iBRB]和外部BRB[oBRB])。与脑血管类似的视网膜血管维持内部血眼屏障。正如BBB或BSCB,这种生理屏障包括紧密连接(TJ)的单层无孔内皮细胞。视网膜上皮细胞间的这些连接阻止大分子从脉络膜毛细血管穿过进入该视网膜。
在2007年,全球总的眼用药物制品方面估值约115.2亿美元(Visiongain,Ophthalmics.2007,Visiongain,Inc.:SanFrancisco.JanoriaKG,等人。Novelapproachestoretinaldrugdelivery.ExpertOpinDrugDeliv。2007;4(4):371-388)。视网膜疾病包括色素性视网膜炎、黄斑变性、视锥-视杆细胞营养不良(CORD)、视网膜脱离、视网膜剥落、高血压性视网膜病变和糖尿病视网膜病变、视网膜母细胞瘤、视网膜脂血症等。
由于许多群体的年龄不断增加,金融专家预测,短期内该方面的年均增长将超过10%。然而,持续给药治疗上有效浓度的药物以治疗眼疾,尤其是眼后半段疾病,仍然是显著的技术挑战。患者顺应性与现有疗法的相应问题也是严重的难题。因此,对于追求新型分子以及创新的药物递送体系的公司而言,存在大量的市场机会。
因此,BBB、BSCB和BRB不仅代表了在使用潜在药物治疗许多CNS和眼部疾病时的主要障碍,还代表了在血液和神经组织之间大面积的潜在交换。
作为一般规律,仅有大约450至600道尔顿的少许亲脂性小分子(仅2%的候选药物)可穿过上述生理屏障,也就是说,穿过血液进入神经组织。许多在治疗CNS疾病的体外研究和动物研究中显示出有希望的结果的候选药物的分子量和大小是相当大的。因此,由于神经组织毛细血管内皮细胞的低跨细胞渗透性,大多数的分子,例如治疗性的肽、蛋白,包括治疗性的抗体,通常被排除在血液到CNS或眼(视网膜)神经组织的通路/转运系统外。排列在血管中的脑毛细血管内皮细胞(BCEC)被基底层、星形胶质细胞突触小结、周细胞以及小神经胶质和神经细胞所包围。内皮细胞与星形胶质细胞突触小结的紧密结合导致了BBB对大多数分子不透过性的发展与维持,从而确保严格且有效地控制血液和脑之间的分子交换以维持脑稳态。相比于其他器官的具有孔的内皮细胞,脑的内皮细胞是通过TJ而紧密结合的。因此这些TJ防止穿过BBB的细胞旁通道。围绕在其周围的内皮细胞和星形胶质细胞突触小结也构成了生理屏障,因为这些细胞具有有效的外排体系,其限制任何跨细胞途径的通路/转运系统。事实上,多药耐药性(MDR)转运蛋白将一些能够穿过生理屏障的分子从内皮细胞主动排出到血液系统。这些主动外排转运(AET)体系通常控制由神经组织到血液系统的小分子的主动外排。举个例子,BBB的模型AET体系是ATP结合盒(ABC)转运蛋白,即P-糖蛋白(P-gp);但是,其他的AET体系也存在于BBB,例如MDR-相关蛋白1(MRP1)。主要位于BCEC腔表面的P-gp不仅是阻止大多数外源物进入脑的BBB生理屏障功能的主要要素,还是候选药物以及在CNS中能够被活化的感兴趣的其他治疗分子。因此,这些性质强烈地限制了物质从血浆转运到CNS和眼神经组织的细胞外隙。
可以解释为什么目前没有可用于主要CNS或眼部病症和损伤(脑癌、帕金森病和阿尔茨海默病、脑血管意外(CVA)等)的真正有效的治疗的原因之一是:研发治疗脑部病症的候选药物的人员采取的是自身研究项目(脑部药物开发项目),而并没有投入精力在穿透屏障的问题,也没有投入精力在优先靶向神经组织,特别是脑(脑部药物靶向项目),(Pardridge,2003,Mol.Interv.,3,90-105)。候选药物必须遵循一定结构、物理化学、药物化学和药理学规律以便具有最大的成为治疗CNS病症或障碍的药物的可能性(Pajouhesh等人,2005,NeuroRx,2(4),541-553)。因此,在研发候选药物中,分子对其靶点的选择性与特异性(药理学性质)对其治疗活性(有效性)是必要的。分子的生物利用度和潜在毒性(药物学性质)对其将来作为药物是至关重要的。换句话说,任何可能成为用于治疗CNS或眼部病症或障碍的药物的分子必须穿过BBB/BSCB或BRB,保持其生物活性并呈现出合适的药代动力学(PK)、吸收、分布、代谢和排泄/清除(ADME)和药效学(PD)特性,且具有低毒性(Tox)。实际上,对于神经系统治疗学这一领域的药学化学家而言,找出正研发的分子的亲水/亲油平衡是特别困难的。
因此,在治疗CNS和眼部障碍和病症中一个主要的问题在于以下事实:所给药的分子不穿过BBB/BSCB/BRB,因此无法到达它们在CNS或眼中的靶点。因此,在发现治疗、诊断或成像CNS或眼部障碍或病症的分子的研究中,一个优先考虑事项是找到提高活性物质穿过BBB/BSCB或BRB的有效性的方法。
在这方面,负载分子穿过这些屏障的策略,即候选药物研发者为了使感兴趣的治疗分子到达CNS而在目前所研究并使用的,可分为两大主要策略:药理学方法和生理学方法(Pardridge,2007,Pharm.Res.,24(9),1733-1744;DeBoer等人,2007,Clin.Pharmacokinet.,46(7),553-576;DeBoer等人,2007,Annu.Rev.Pharmacol.Toxicol.,47,327-355;Jones等人,2007,Pharm.Res.,24(9),1759-1771;Vlieghe和Khrestchatisky,2012,MedResRev.,33(3),457-516)。
侵入方法
侵入方法可通过以下实施:在脑中直接室内注射、脑内注射或鞘内输注活性物质或破坏BBB/BSCB(短暂的破坏这些屏障的完整性)。
对于通过室内注射的神经外科方法,除了涉及神经外科手术的花费之外,其主要问题在于:该药物不是直接递送至脑实质而是递送至脑脊液。室内输注包括将导管置于室(Aird,1984,Exp.Neurol.,86,342-358)。这个高度侵入性技术无法有效地将活性物质输送到神经实质。事实上,例如在脑中,在通过室内输注递送药物的过程中,脑脊液到脑实质的流量是由其对流(输送)的异常缓慢的扩散所控制的,因为脑不具有实质内流量。
同样对于脑内注射,脑中活性物质的扩散从注射部位到病变部位降低得非常快。事实上,活性物质的脑浓度在距离其注射部位500μm处减少了90%。
鞘内输注包括将导管置于脑中。这个导管被连接至以预定流速递送活性物质的泵。由于脑是唯一不具有淋巴系统的器官这一事实,而淋巴系统通常提供将细胞外液输送回大循环,因此通过鞘内输注将活性物质分布于脑中是非常缓慢的。这降低了活性物质在病变部位的浓度。
此外,在该神经外科手术过程中,感染的风险显著,特别是在所述导管的存在的情况下。在这些条件下,患者舒适性不佳。
BBB的短暂开放与BCEC的瞬间开放TJ有关。这是针对血管活性物质,例如白细胞三烯或缓激肽的情况下(Baba等人,1991,J.Cereb.BloodFlowMetab.,11,638-643)。这一策略同样是侵入性的且需要动脉进入麻醉了的受试者/患者的颈动脉。短暂破坏BBB的完整性遇到的主要问题,除了涉及进入颈动脉的放射性手术的花费外,所述BBB仅维持很短时间的开放,因此限制了长期递送药物的可能性。此外,短暂破坏BBB会使血浆蛋白进入脑(而这些蛋白可能对脑有毒性)且还可促使病原体的进入。因此,破坏BBB的这种类型可能会导致慢性神经病理性破坏并伴随高风险感染(Salahuddin等人,1988,ActaNeuropathol.,76,1-10)。
负载的药理学方法
输送分子的药理学方法包括:通过将脂质或亲脂性基团加至活性物质中所制成的更疏水分子的跨细胞扩散(跨细胞亲脂性扩散,TLD)或使用脂质体(Zhou等人,1992,J.Control.Release,19,459-486),以及通过正电荷载体分子的离子吸附或通过将活性分子阳离子化进行输送(吸附介导的输送或AMT)。
加入脂质或亲脂性基团能够使亲水性分子化学转化成更疏水的分子,尤其是利用前药方法。但是,合成该化合物导致超出最佳输送阈的分子穿过BBB/BSCB/BRB,尤其是对于分子量(MW)变得大于450道尔顿的最佳限度(Pajouhesh等人,2005,NeuroRx,2(4),541-553)。出于相同原因,通常过大的脂质体或甚至小囊泡(胶束等)或纳米颗粒(纳米球,纳米囊)对BBB/BSCB/BRB没有足够的特异性,并因此不太能够有效地输送感兴趣的治疗分子(或成像或诊断试剂或任何其他分子,例如分子探针)穿过这些屏障(Levin,1980,J.Med.Chem.,23,682-684;Schackert等人,1989,SelectiveCancerTher.,5,73-79)。此外,这种类型的囊泡系统通常对大脑具有不可忽略的毒性作用。因此,脂质化技术所面临的主要问题是相比于其他细胞膜,它们对特异性靶向并穿过BBB/BSCB/BRB的特异性较低、该药物血浆值的曲线下面积(AUC)的降低以及它们普遍仅限用于负载小分子。
在AMT(通过共价结合或直接阳离子化药物引入阳离子基团)中,所面临的主要问题是相比于其他细胞膜,特异性靶向和穿过BBB/BSCB/BRB的特异性低。事实上,AMT基于吸附在细胞上的阳离子分子,所述细胞的细胞膜带负电荷,这是对于绝大多数细胞而言。该药物AUC的血浆值的降低、其通常仅限用于负载小分子以及它们的细胞毒性均是使AMT负载方法无效的其他因素。
负载的生理学方法
基于生理学方法进行负载的策略在于利用屏障的各种天然输送机制。主动输送分子穿过BBB的这些机制既可以通过偶联特定受体底物或与特定受体底物分子模拟起作用(载体介导的输送或CMT),也可以通过与特异性靶向受体的配体偶联或融合起作用(RMT,受体介导的转运或受体介导的跨细胞作用)。
作为一个例子,分子(例如L-DOPA(帕金森病)、美法仑(脑癌)、α-甲基-DOPA(动脉高血压)和加巴喷丁(癫痫))通过CMT,经由大型中性氨基酸转运蛋白1和2(LAT1和LAT2)进入脑(Pardridge,2003,Mol.Interv.,3,90-105)。这些分子具有与苯丙氨酸(LAT1天然底物之一)接近的化学结构。但是,CMT方法面临的主要问题是它们对缀合物广泛的选择性/特异性,所述缀合物近似地模仿/模拟内源性受体/转运蛋白的底物,并因此它们的用途仍然仅限于负载小分子。
RMT需要受体依赖的输送系统。负载是经由胞吞作用机制,通过靶向存在于靶组织(包括脑毛细血管)中的内源性受体实现的。各种会参与RMT的人BBB受体的著名实例包括:转铁蛋白受体(TfR),其输送结合铁的转铁蛋白(Tf);胰岛素受体(IR)或胰岛素样生长因子受体(IGFR);能够输送包含在低密度、高密度和极低密度脂蛋白(分别是LDL、HDL和VLDL)中胆固醇的受体,包括低密度脂蛋白(LDL)受体和低密度脂蛋白受体相关蛋白(LRP)家族成员;白喉毒素受体(DTR)或肝素结合性表皮生长因子(HB-EGF);以及清道夫受体(SCAV-Rs),包括B族I型清道夫受体(SR-BI)。在RMT中,器官中特定细胞(包括BBB内皮细胞)细胞膜上的受体结合它们的配体,其导致由在细胞表面形成的囊泡中受体及其配体所组成的缀合物的胞吞作用,然后穿透靶细胞。这个RMT过程并不依赖于胞吞作用所吞食的分子大小。对于BBB/BSCB/BRB,所述配体/受体缀合物或离解配体可穿过内皮细胞(跨细胞作用),并因此能够穿过这些生理屏障,从而作用于神经组织。因此,许多分子或分子缀合物的RMT是一种将其输送至特定细胞类型或器官,尤其是从血液到CNS或眼的机制。这些分子或分子缀合物的受体可用于输送这些经修饰而携带药物的受体的天然配体和/或纳米颗粒。例如,将纳米颗粒用聚山梨酯,尤其是聚山梨酯80涂布会导致载脂蛋白E从血浆中吸收至该纳米颗粒的表面,随后该纳米颗粒模仿低密度脂蛋白(LDL)颗粒且可与LDLR相互作用,从而导致其被内皮细胞所摄取(Kreuter,2012,AdvDrugDelivRev.pii:S0169-409X(12)00275-X)。LDL受体靶向的包裹有脂质体的多柔比星提高了跨越BBB细胞的体外药物递送(Pinzón-Daza等人,2012,BrJPharmacol。doi:10.1111/j.1476-5381.2012.02103.x)。Tf是存在于BBB上的TfR的天然配体。已将活性物质偶联/缀合至Tf,从而通过Tfr来主动穿透BBB(Jefferies等人,1984,Nature,312,162-163;Friden等人,1983,Science,259,373-377;Friden,1994,Neurosurgery,35,294-298)。尽管使用蛋白型大分子的这个负载策略能够提高感兴趣的缀合分子穿透该屏障,但是该策略有多个缺点。首先,该分子通常经基因表达方法偶联/缀合至载体(融合),因此将所输送的分子数仅限制为多肽或蛋白。第二,用于偶联/缀合分子和载体的系统是非常复杂的;传统的化学或生化偶联无法从结构和分子角度得到明确的大分子体系。此外,缀合物和靶向受体的内源性配体之间的潜在竞争可导致RMT生理过程的抑制,或导致脑正常运转所需的内源性配体的浓度降低。最后,RMT受体还可参与大脑的细胞信号传导过程且所述缀合物可能会干扰这些过程。
经特异性受体的RMT还可用于靶向药物至其他组织/器官而非BBB和脑。已显示,例如视网膜对胆固醇的摄取主要经LDLR介导的过程发生(Tserentsoodol等人,2006,MolecularVision2006;12:1306-18)。
器官的血管系统、具体器官的实质或患病组织可表达高水平的给定受体,该受体可用于药物靶向(reviewedinChung和Wasan,2004,AdvDrugDelivRev.2004May7;56(9):1315-34)。例如LDLR在肝(尤其是在肝细胞窦状隙侧)以及在其他组织,例如肾上腺和肠中高水平表达(Beisiegel等人,1981,JBiolChem.25;256(8):4071-8;Huettinger等人,1984,JClinInvest.;74(3):1017-26;Fong等人,1989,JClinInvest;84(3):847-56)。
最终,有足够的证据显示了在各种类型的癌症中LDLR增加的表达并显示了经由这些癌细胞(包括胶质母细胞瘤)的LDLR进行的LDL-或纳米颗粒介导的抗癌药物的递送(Varshosaz等人,2012,EurJMedChem;54:429-38;Kopecka等人,2011;JournalofControlledRelease;149:196–205;Nikanjam等人,2007,IntJPharm.;328(1):86-94;reviewedinNg等人,2011,AccChemRes;44(10):1105-13;Firestone,1994,BioconjugChem.;5(2):105-13.)。
还已经表明,在各种LDLR内源性配体(例如LDL或PCSK9)结合至LDLR后,所述内源性配体要经历受体介导的胞吞作用,随后经历膜囊(核内体)的细胞内运输,最终与溶酶体融合(Issandou等人,2004,BiochemPharmacol.;67(12):2281-9;reviewedinLambert等人,2009,Atherosclerosis.;203(1):1-7)。
溶酶体贮积病(LSD)代表了约70种遗传学上不同的病症,伴有合并的出生缺陷率约为1/7500。酶替代疗法(ERT)需要摄取用于治疗多发性LSD的重组酶。甘露糖-磷酸受体(M6PR)是目前酶递送至组织以及递送至细胞溶酶体区室的主要靶点(ReviewedinCox,2012,JPathol.;226(2):241-54;Lachmann,2011,CurrOpinPediatr.;23(6):588-93),但其他受体,例如那些涉及RMT的受体也可以被考虑,尤其是在靶向ERT至CNS中,认为大约2/3的LSD会影响CNS。
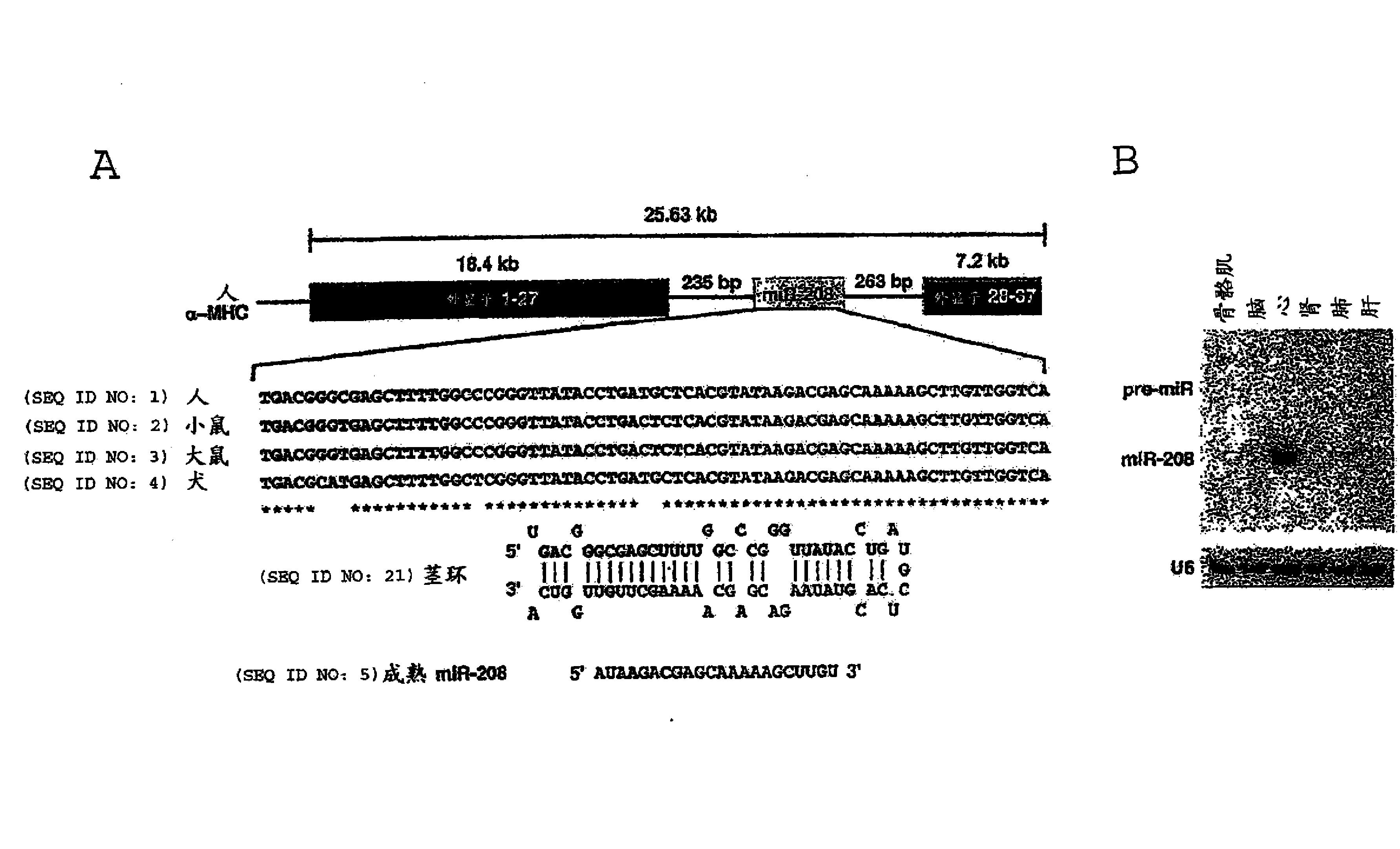
国际专利申请WO2010/046588首次描述了肽或伪肽,其结合人、小鼠或大鼠LDLR且能够携带可以是高分子量和/或大体积的物质穿过BBB。
本申请涉及结合LDLR,尤其是人、小鼠和/或大鼠LDLR的新型肽,且所述肽被优化用于将分子定位至表达LDLR的健康或病理(癌细胞)的组织(例如肝、肾上腺,肠),尤其是穿过神经系统的生理屏障,例如BBB、BSCB或BRB。
发明内容
本发明提供新型肽或伪肽,其能够输送感兴趣分子穿过表达LDLR的组织的细胞膜,尤其是穿过CNS或视网膜的生理屏障(BBB/BSCB和BRB)。因此,本发明使设计新型治疗剂或诊断剂成为可能,所述治疗剂或诊断剂具有提高的生物分布或生物利用度,尤其是对CNS和/或眼和/或富集LDLR的其他器官(例如肝、肾上腺和肠)和/或富集LDLR的不同癌细胞和/或细胞的溶酶体区室具有改进的穿透(靶向)。
在WO2010/046588和WO2011/131896中,发明人已经研发了能够结合LDLR并能够定向分子(包括感兴趣的治疗或诊断分子)至脑的肽衍生物。
发明人目前已经设计并确定了该新型肽,其呈现出输送分子的有利性质。该发明人还设计了呈现出提高的亲和力以及递送任意药剂至动物的高输送能力的肽的新型多聚体。这些新型肽和多聚体无需与天然配体竞争就能够结合LDLR,且因此不干扰内源性LDL的输送,且所述新型肽和多聚体代表新型产物,所述产物特别有利于递送、靶向或负载治疗性或诊断性(包括成像)药物或试剂,尤其是递送至CNS和/或眼和/或富集LDLR的其他器官(例如肝、肾上腺和肠)和/或富集LDLR的癌细胞和/或细胞的溶酶体区室。
本发明的一个方面具体地涉及包含选自SEQIDNO:1-5序列的肽或伪肽。
本发明的另一个方面是下式(A)的多聚体试剂(multimericagent):
Ph-M-Pi(A)
其中,
各P代表相同或不同的肽或伪肽,其包含氨基酸序列A1-Met-A2-Arg-Leu-Arg-A3-A4,其中A1和A4独立地表示半胱氨酸或其类似物或其电子等排体,A2表示脯氨酸或其类似物或其电子等排体,且A3表示甘氨酸或其类似物或其电子等排体;
M是分子平台(Molecularplatform),且
h和i是彼此独立地选自1、2、3、4或5的整数。
根据优选的实施方案,A1表示半胱氨酸(Cys)或其类似物,其选自:(D)-半胱氨酸、青霉胺(Pen)和(D)-青霉胺((D)-Pen);A2表示脯氨酸(Pro)或其类似物,其选自:哌可酸(Pip)和噻唑烷-4-羧酸(Thz);和/或A3表示甘氨酸(Gly)或肌氨酸(Sar)和/或A4表示半胱氨酸或其类似物,其选自:(D)-半胱氨酸、青霉胺(Pen)和(D)-青霉胺((D)-Pen)。
在优选的实施方案中,P是具有以下通式(I′)的肽或伪肽:
(D)-Cys-Met-A2-Arg-Leu-Arg-A3-A4(I′)
其中A2表示脯氨酸或其类似物,其优选地选自Pip和Thz;A3表示甘氨酸(Gly)或肌氨酸(Sar)和/或A4表示半胱氨酸或其类似物,其选自:(D)-半胱氨酸、青霉胺(Pen)和(D)-青霉胺((D)-Pen)。
在最优选的实施方案中,P是包含选自SEQIDNO:1-5中任一个序列的肽。
在最优选的实施方案中,h=i=1。
本发明的肽和多聚体有利地具有以高亲和力结合人LDL受体(hLDLR)、小鼠LDLR或大鼠LDLR的能力且具有输送感兴趣分子至表达LDLR的组织和细胞或穿过表达LDLR的细胞(例如内皮细胞)的能力。
在这一方面,本发明的另一方面是如上所述结合至一或多个分子,优选一或多个诊断性或治疗性分子的肽或多聚体。
本发明的另一方面涉及如上所定义的肽或伪肽或多聚体在哺乳动物中递送或输送治疗性或诊断性试剂中的用途。
本发明另一方面涉及例如上文所定义的用于在哺乳动物中递送或输送治疗性或预防性试剂的肽或伪肽或多聚体。
本发明的其他方面涉及例如上文所定义的用于制备药物或诊断组合物的肽或伪肽或多聚体的用途。
本发明的另一方面涉及例如上文所定义的用于提高生物活性或降低活性物质或其所偶联的感兴趣物质的毒性的肽或伪肽或多聚体的用途。
本发明的另一方面涉及具有下式(II)的任意接合化合物(conjugatedcompound):
VxDy(II)
其中V表示例如上文所定义的肽或伪肽或多聚体,D表示活性物质或感兴趣的物质,且x和y是1至5的整数。
本发明还涉及具有下式(III)的任意接合化合物:
VxSzDy(III)
其中V表示例如上文所定义的肽或伪肽或多聚体,S表示间隔基(spacer),D表示活性物质或感兴趣的物质,x是1至5的整数且y和z是1至10的整数。
本发明的另一方面涉及药物组合物,其包含结合至例如上文所定义的肽或伪肽或多聚体的活性成分和一种或多种药学上可接受的赋形剂。
本发明的另一方面涉及诊断性组合物,其包含结合至例如上文所定义的肽或伪肽或多聚体的诊断或成像试剂。
本发明的另一方面涉及改善或促进分子穿过表达LDLR的健康或病理的组织的细胞膜,且尤其是穿过生理屏障BBB/BSCB和BRB的方法,其包括将所述分子偶联至例如上文所定义的肽或伪肽或多聚体。
本发明的另一方面是使用药物来治疗受试者病症的改进的方法,所述改进包括将所述药物偶联至例如上文所定义的肽或伪肽或多聚体。
本发明可用于任意动物,具体是任意哺乳动物,且更具体是用于人。
附图说明
图1.以直接连接合成以及通过治疗感兴趣的肽/分子缀合物的连接基团合成的比较图。
图2.与报道分子或荧光分子(例如生物素、荧光素(FITC)、若丹明(若丹明RED-X)、青色素(Cy3.5)或Stag肽)经C端(C-term)间隔基/连接基团结合的合成肽的总线路图。
图3.使用在盖玻片上生长并用DiI-LDL20mg/mL和肽-STag缀合物(SEQIDNO:1)10μM一起培养的CHO-hLDLR-GFP细胞进行的脉冲追踪实验。在4℃培养30分钟之后,将细胞充分洗涤,然后在追踪培养基中培养5分钟或3小时,使进行结合肽的胞吞作用和细胞内运输。细胞核使用烟酸己可碱(Hoechst)染色。插图显示了高倍放大的共焦成像(confocalimage)的代表区域,并且该插图表明了在所述肽与LDL运输(T5)早期步骤中,所述肽与LDLR和LDL的共区域化,以及在不含LDLR的含LDL的溶酶体中的肽的后积累(lateaccumulation)(T180–插图2)或在不含LDL的含LDLR的回收小囊泡中的肽的后积累(T180–插图1)。
图4.人IgG1抗体的Fc片段被用作原型蛋白部分,其经由化学连接至SEQIDNO:1的肽载体输送至细胞。与参照肽2(称作“SEQIDNO:43”,是在WO2010/046588中公开为SEQIDNO:43的氨基酸序列)的融合,其通过重组分子技术制备,用于比较目的。脉冲追踪实验显示了Fc-SEQIDNO:1与CHO-hLDLR-GFP细胞的结合(化学连接)以及Fc-SEQIDNO:43与CHO-hLDLR-GFP细胞的结合(融合)。
将在盖玻片上生长的CHO-hLDLR-GFP细胞与5nM的化学连接的Fc-SEQIDNO:1、融合蛋白Fc-SEQIDNO:43或单独的Fc在4℃培养30分钟,充分洗涤,然后在37℃追踪培养基中培养5分钟,使进行结合蛋白的胞吞作用。细胞核使用烟酸己可碱染色。Fc用结合至Alexa594的抗人Fc抗体进行检测。请注意,仅Fc-SEQIDNO:1和Fc-SEQIDNO:43结合hLDLR。插图显示了高倍放大的共焦成像的代表区域,并且该插图表明了LDLR和Fc-肽缀合物在早期/拣选核内体中局部化的区域。
图5.体外血脑屏障(BBB)模型:与星形胶质细胞共培养的大鼠脑毛细血管内皮细胞(RBCEC)。在A中,将活的RBCEC用40μg/mL的DiI-LDL培养30min。在B中,将标记在活RBCEC表面的大鼠LDLR与识别人、小鼠和大鼠LDLR的胞外域的山羊抗体在10μg/mL培养1h。在C,将相同抗体标记大鼠RBCECLDLR,然后用PFA4%固定细胞。
流式细胞术(FACS):RBCEC单层与EDTA0.25mM的机械分离。在D中,对照RBCEC被藻红蛋白PE-A标记。在E中,在4℃,DiI-LDL以40μg/mL结合至RBCEC的LDLR受体,持续90min。在F中,在4℃,DiI-LDL以40μg/mL结合至LDLR-/-大鼠(LDLR敲除,KO大鼠)的RBCEC,持续90min。在G中,仅用二抗(驴抗山羊别藻蓝蛋白APC,用作对照)的RBCEC。在H中,在4℃,用10μg/mL的山羊抗LDLR抗体标记45min,然后用二抗标记。在I中,在4℃,将10μg/mL的山羊抗LDLR抗体标记在LDLR-/-大鼠的RBCEC上,持续45min,然后用二抗标记。在J中,体外BBB模型:将大鼠脑毛细血管内皮细胞(RBCEC)与星形胶质细胞共培养:在活的WT和LDLR-/-RBCEC中在培养30min后在不同浓度的低区室DiI-LDL中的输送。
图6.体外BBB模型:大鼠脑毛细血管内皮细胞(RBCEC)与星形胶质细胞共培养。A至F:在野生型(WT)大鼠的活的RBCEC中的培养,G和H:LDLR-/-大鼠的活的RBCEC。在A中,在活的WTRBCEC中培养30min,结合/摄取0.25μM的Fc。在B中,在活的WTRBCEC中培养30min,结合/摄取0.25μM的Fc-SEQIDNO:43。在C和E中,在活的WTRBCEC中培养30min,结合/摄取40μg/mL的DiI-LDL。在D中,在活的WTRBCEC中培养30min,结合/摄取0.25μM的Fc-SEQIDNO:43。这些显微照片显示了在DiI-LDL和Fc-SEQIDNO:43之间的共区域化。在E中,在LDLR-/-大鼠活的RBCEC中培养30min,结合/摄取40μg/mL的DiI-LDL。在F中,在LDLR-/-大鼠活的RBCEC中培养30min,结合/摄取0.25μM的Fc-SEQIDNO:43。在G中,在活的WTRBCEC中培养30min,结合/摄取0.1μM的Fc-CTRL。在H中,在LDLR-/-大鼠活的RBCEC中培养30min,结合/摄取0.1μM的Fc-SEQIDNO:1。
图7.体外BBB模型:将大鼠脑内皮细胞(RBCEC)与星形胶质细胞共培养。
在A中,在活的WTRBCEC中培养30min后,结合/摄取不同浓度的Fc和Fc-SEQIDNO:43。在B中,在活的WTRBCEC中培养2h后,结合/摄取0.1μM的Fc-SEQIDNO:1和Fc-CTRL。
图8.肽载体二聚体。二聚体比其单体形式显示出了对LDLR更好的亲和力。
在A中,两个二聚体实例是基于两种不同的分子结构。A-1:该二聚体实例是经三胺市售平台上的肽偶联合成的。R可以是氢、示踪分子、活性物质或在之间有无连接基团的任意感兴趣的物质。A-2:该二聚体实例是经“自制”二-赖氨酸平台上的肽偶联合成的。R1和/或R2可以是氢、示踪分子、活性物质或在之间有无连接基团的任意感兴趣的物质。
在B中,通过ELISA抗-STag定量肽SEQIDNO:1-STag于CHO-hLDLR-GFP的结合/摄取。在37℃,将CHO-hLDLR-GFP细胞单独地用10μM的肽SEQIDNO:1-STag培养60min(100%结合)或用10μM与肽SEQIDNO:1、肽CTRL或肽D2(SEQIDNO:1)2竞争的肽SEQIDNO:1-STag培养60min。
在C中,在4℃,经30min,将500nM的肽D2(SEQIDNO:1)2-Cy3.5和D2(CTRL)2-Cy3.5结合至CHO-hLDLR-GFP。将细胞核用烟酸己可碱染色。显微照片显示了肽D2(SEQIDNO:1)2-Cy3.5和LDLR在质膜上的共区域化。
图9.在WT小鼠或LDLR-/-小鼠(KO)中,单独地肽SEQIDNO:1或化学偶联至人Fc片段的SEQIDNO:1的组织分布。
在A中,在WT或LDLR-/-小鼠尾静脉注射后10min的氚标记的肽SEQIDNO:1的组织分布。结果表示为每克组织的总注射剂量百分比(%DI/g)。组织中放射性的计数显示,肽在LDLR富集的WT组织中比在LDLR-/-小鼠中积累的多。在B中,在WT或LDLR-/-小鼠尾静脉注射后2小时的化学偶联的Fc-SEQIDNO:1的组织分布。结果表示为每克组织的总注射剂量百分比(%DI/g)。使用ELISA测试对组织中Fc部分的积累进行的定量显示,Fc在LDLR富集的WT组织中比在LDLR-/-小鼠中积累的多。
发明详述
本发明涉及能够结合人LDLR的肽衍生物以及其在药物制剂领域中的用途,尤其是在输送感兴趣的治疗或诊断分子穿过健康或病理(例如,癌细胞)的表达LDLR的组织(例如肝、肾上腺或肠)的细胞膜,且尤其是穿过生理屏障BBB/BSCB和BRB中的用途。本发明的肽和多聚体对人的LDLR具有高亲和性且可在体内输送分子。本发明的肽或伪肽和多聚体可容易地被化学合成,且感兴趣的任意治疗或诊断(例如,成像)分子可经由间隔基简单且有效地进行偶联(经由连接基团合成)或通过在两个实体之间直接偶联(直接连接合成)(图1)。肽和伪肽被设计成环状结构,因此对蛋白水解具有更好的抗性。此外,本发明的肽和多聚体结合LDLR而不与天然配体竞争。
本发明的肽或伪肽或多聚体可在治疗、成像和/或诊断神经病症以及脑或其他组织的遗传、感染、炎性或癌性病症中用作感兴趣的治疗分子、或成像剂或诊断剂、或任何其他分子(例如分子探针)的载体。
本发明所述的肽、伪肽和多聚体具有靶向细胞受体和特定细胞类型和器官,尤其是癌细胞、神经或非神经组织(例如肝、肾上腺或肠)的能力,和/或具有穿透细胞膜的能力,尤其是神经系统(更特别的是CNS和眼)的那些生理屏障,特别是BBB、BSCB、BRB或癌性神经组织肿瘤的血-瘤屏障(BTB)。
本发明的肽、伪肽和多聚体具有结合人、小鼠和/或大鼠特定细胞类型或器官(例如肝、肾上腺或肠)的细胞膜LDLR的能力,尤其是具有结合CNS和眼的生理屏障LDLR的能力,且具有通过胞吞作用或受体介导的输送/跨细胞作用(RMT),经由这一受体穿过上述膜的能力。
因此,肽、伪肽和多聚体可用于设计适用于治疗各种疾病(例如感染性病症或细菌、病毒、寄生虫或真菌性质/来源的其他病症)的药物。
本发明的一个方面尤其涉及包含选自SEQIDNO:1-5的序列的肽或伪肽。
SEQIDNO:1,(D)-Cys-Met-Thz-Arg-Leu-Arg-Gly-Pen;
SEQIDNO:2,(D)-Cys-Met-Thz-Arg-Leu-Arg-Sar-Pen;
SEQIDNO:3,(D)-Cys-Met-Pip-Arg-Leu-Arg-Sar-Cys;
SEQIDNO:4,(D)-Cys-Met-Pip-Arg-Leu-Arg-Gly-Pen;
SEQIDNO:5,(D)-Cys-Met-Pip-Arg-Leu-Arg-Sar-Pen。
本发明的其他方面涉及具有下式(A)的多聚体试剂:
Ph-M-Pi(A)
其中,
各P表示相同或不同的肽或伪肽,其包含氨基酸序列:A1-Met-A2-Arg-Leu-Arg-A3-A4,其中A1和A4独立地表示半胱氨酸或其类似物或其电子等排体,A2表示脯氨酸或其类似物或其电子等排体,且A3表示甘氨酸或其类似物或其电子等排体;
M是分子平台,和
h和i是彼此独立地选自1、2、3、4或5的整数。
A1通常表示选自以下的残基:D或L构型的半胱氨酸(Cys,C)或其衍生物,该衍生物选自2-氨基-3-巯基丙酸和其S-取代的衍生物;S-乙酰基半胱氨酸或2-氨基-3-(乙酰基硫基)丙酸;硒基半胱氨酸(Sec,U)或2-氨基-3-(硒基)丙酸;半胱氨醇;3-巯基丙酸(Mpa);或L或D构型的青霉胺(Pen)。在优选的实施方案中,A1为D-Cys、Pen或D-Pen。
A2优选地表示选自以下的残基:脯氨酸(Pro,P)或吡咯烷-2-羧酸、高脯氨酸或2-(2-吡咯烷基)乙酸、3-羟基脯氨酸(3Hyp)、4-羟基脯氨酸(4Hyp)、3-甲基脯氨酸、3,4-二氢脯氨酸、3,4-亚甲基脯氨酸、4-氨基脯氨酸、4-氧代脯氨酸、硫代脯氨酸或噻唑烷-4-羧酸(Thz)、2-氧代噻唑烷-4-羧酸、吲哚-2-羧酸(Idc)、哌可酸(Pip)或哌啶-2-羧酸、哌啶酸(Nip)或哌啶-3-羧酸、4-氧代哌可酸、4-羟基哌可酸、氨基-1-环己烷羧酸、脯氨醇。在优选的实施方案中,A2选自Pro、Pip或Thz。
A3优选地表示选自以下的残基:甘氨酸(Gly,G)或2-氨基乙酸、肌氨酸(Sar)或N-甲基甘氨酸(MeGly)、N-乙基甘氨酸(EtGly)、烯丙基甘氨酸(allylGly)或2-氨基戊-4-烯酸、2-环戊基甘氨酸(Cpg)、2-环己基甘氨酸(Chg)、2,2-二丙基甘氨酸(Dpg)、2-(3-吲哚基)甘氨酸(IndGly)、2-茚满基甘氨酸(Igl)、2-新戊基甘氨酸(NptGly)、2-辛基甘氨酸(OctGly)、2-炔丙基甘氨酸(Pra)或2-氨基戊-4-炔酸、2-苯基甘氨酸(Phg)、2-(4-氯苯基)甘氨酸、氮杂甘氨酸(AzGly)或甘氨醇或2-氨基乙醇。在优选的实施方案中,A3选自Gly和Sar。
A4优选地表示选自以下的残基:D或L构型的半胱氨酸(Cys,C)或其衍生物,该衍生物选自2-氨基-3-巯基丙酸和其S-取代的衍生物;S-乙酰基半胱氨酸或2-氨基-3-(乙酰基硫代)丙酸;硒基半胱氨酸(Sec,U)或2-氨基-3-(硒基)丙酸;半胱氨醇;或L或D构型的青霉胺(Pen)。在优选的实施方案中,A4选自D-Cys、Pen或D-Pen。
在本发明优选的一类多聚体试剂中,P代表式(I′)的肽或伪肽:
(D)-Cys-Met-A2-Arg-Leu-Arg-A3-A4(I′)
其中A2、A3和A4如上所定义。
P基团的具体实例为包含SEQIDNO:1-5中任一个的肽。
本发明的多聚体试剂可包含2-10个P基团,其可以是相同或不同的。在优选的实施方案中,所述多聚体试剂是同源多聚体,即,所有的P基团都是相同的。此外,本发明优选的多聚体试剂是二聚体或三聚体,更优选为二聚体。本发明确实显示,通过使用二聚体形式的上述LDLR-结合的肽,该结合能力得到改善且RMT性质得到提高。现有技术无法预期该结果,因为该受体的天然配体是单体。
在一个具体的实施方案中,在本发明的多聚体试剂中,h是1或2而i是1或2。在更优选的实施方案中,h=i=1。
分子平台M可以是任意适用于药物或兽医领域的化学交联基团。该基团优选地不具有生物活性和毒性。该分子平台的大小可由本领域技术人员进行调整。M基团的优选实例是聚赖氨酸平台或多官能有机化合物,例如三(2-氨基乙基)胺。其他M基团为市售可得的有机化合物。在一个具体的实施方案中,所述分子平台含有至少两个反应官能团,优选至少三个,从而偶联至少2个肽和至少一种感兴趣的分子。应当注意,感兴趣的分子可被偶联至肽或该平台。反应官能团的实例包括,例如,胺、酸、硫醇、叠氮化物、炔烃、羰基化合物或肼。在优选的实施方案中,所述分子平台是聚赖氨酸基团,优选包含2-10个赖氨酸。具体的实例是二赖氨酸或含有相同数量的赖氨酸残基作为肽P的聚赖氨酸。所述肽可通过与该平台的反应能团的共价键直接偶联至该平台,或通过间隔基偶联至该平台,所述间隔基可由例如甘氨酸或一系列甘氨酸、PEG分子或氨基己酸组成。
多聚体试剂的具体且优选的实例是下式分子:
其中“SEQIDNO:1-5”是指包含选自SEQIDNO:1-5中任一个序列的肽。应理解,上述式中的肽可包含如上所定义的式A1-Met-A2-Arg-Leu-Arg-A3-A4的任意序列。
本申请所获得的结果表明,本发明的肽和多聚体对LDLR具有增加的亲和力。具体地,相比于参考化合物,SEQIDNO:1-5的肽表现出极高的亲和力,如表1(实施例II)所示。此外,本发明令人惊讶地表明,多聚体试剂呈现出增加的受体亲和力和高靶向组织(例如脑)的能力。更具体地,包含2个如上所定义的HDLR-结合肽的二聚体试剂具有极高地靶向组织(例如脑)的能力。
考虑到小尺寸的肽(上述肽含有8个氨基酸),这些结果更加显著,这构成了其在工业用途中的另一优势。
如上所述,本发明的肽或伪肽或多聚体可包括肽、非肽和/或修饰的肽键。在优选的实施方案中,所述肽或伪肽包含至少一个拟肽键(peptidomimeticbond),其优选地选自亚甲基(-CH2-)或亚膦酸根(-PO2-)、仲胺(-NH-)或氧基(-O-)、α-氮杂肽、α-烷基肽、N-烷基肽、膦酰胺酯(phosphonamidate)、缩肽、羟基亚甲基、羟基乙烯、二羟基乙烯、羟基乙胺、逆反肽(retro-inversopeptide)、亚甲氧基、酮亚甲基(cetomethylene)、酯、次膦酸酯、次膦酸、膦酰胺和卡巴类似物(carbaanalogue)。
此外,在一个具体的实施方案中,本发明的肽或伪肽包含分别被酰化或酰胺化或酯化所保护的N-端和/或C-端官能团。
本发明的肽或伪肽可用本领域技术人员所知的任意技术进行合成(化学、生物或基因合成等)。它们可按原样保存或在感兴趣的物质或任意可接受的赋形剂存在下进行配制。
对于化学合成,使用了商品仪器,其可掺入天然和非天然氨基酸,例如D对映异构体和具有不同于天然类似体的疏水性和空间位阻的侧链的残基(所谓的外来,即,非编码氨基酸),或含有一或多个拟肽键的肽序列,所述拟肽键尤其包括亚甲基(-CH2-)或亚膦酸根(-PO2-)、仲胺(-NH-)或氧基(-O-)或N-烷基肽。
在合成过程中,有可能引入各种化学修饰,例如,将脂质(或磷脂)衍生物或脂质体或纳米颗粒的组成置于N端或C端位置或置于侧链上,使能够将本发明的肽或伪肽并入类脂膜,例如由一或多个磷脂层或磷脂双层组成的脂质体的类脂膜,或纳米颗粒的类脂膜。
本发明的肽或其蛋白部分,还可获自编码所述肽或其蛋白部分的核酸序列。本发明还涉及核酸分子,其包含编码例如上文所定义的肽的核酸序列或由所述核酸序列所组成。更具体地,本发明涉及核酸分子,其包含至少一个编码通式(I)的肽的序列。这些核酸序列可以是DNA或RNA,并且可以与对照序列组合,和/或插入到生物表达载体中。
所用的生物表达载体是根据其转染的宿主进行选择的。它可以是,例如,质粒、粘粒、病毒等。本发明尤其涉及这些可用于在宿主细胞中产生本发明肽或其蛋白部分的核酸和生物表达载体。这些生物表达载体可用本领域技术人员熟知的分子生物学和基因工程技术进行制备且所述肽可用本领域技术人员熟知的分子生物学和基因工程技术产生或表达于宿主。
本发明的另一方面涉及例如上文所定义的肽或伪肽或多聚体作为在运输/输送感兴趣的治疗分子,或成像或诊断剂,或任何其他分子的载体中的用途。
本发明还涉及例如上文所定义的肽或伪肽或多聚体在制备能够穿过表达LDLR的健康或病理的组织的细胞膜,且尤其是穿过生理屏障BBB/BSCB和BRB的药物中的用途。
本发明还涉及使得或改善分子穿过表达LDLR的健康或病理(例如,癌细胞)的组织(例如肝、肾上腺或肠)的细胞膜,尤其是穿过生理屏障BBB/BSCB和BRB的方法,其包括将分子偶联至本发明的肽或伪肽或多聚体。
本发明还涉及具有下式(II)的接合化合物:
PxDy(II)
其中P表示肽或伪肽,其包含SEQIDNO:1-5中任一个的氨基酸序列,D表示活性物质或感兴趣的物质,且x和y是1至5的整数。在一个具体的实施方案中,x和y等于1、x大于y或y大于x。
本发明还涉及下式(III)的接合化合物:
VxSzDy(III)
其中V表示本发明的肽或伪肽或多聚体,S表示间隔基,D表示活性物质或感兴趣的物质,x是1至5的整数和y和z是1至10的整数。在一个具体的实施方案中,x=z=y=1或x=z>y或y=z>x或z>x>y。
活性物质或感兴趣物质可以是感兴趣的任意制药分子,尤其是治疗、诊断或医学成像试剂,或分子探针。它具体地可以是感兴趣的任何生物学化学个体,例如化学小分子(抗生素、抗病毒剂、免疫调节剂、抗肿瘤药、抗炎药等)、肽或多肽(例如神经肽、激素)、蛋白(酶,尤其是溶酶体、激素、细胞因子、载脂蛋白、生长因子、抗原、抗体或抗体的一部分)、核酸(核糖核酸、siRNA、miRNA或人、病毒、动物、真核生物或原核生物的脱氧核糖核酸、植物或合成来源等等,其大小范围可以为从单一寡核苷酸的大小至基因组或基因组片段的大小)、病毒基因组或质粒、核酶、标记物或示踪物。通常,“感兴趣的物质”可以是任意药物活性成分,无论是化学、生物化学、天然或合成的化合物。表述“化学小分子”是指最大分子量为1000道尔顿的感兴趣的药学分子,通常为300道尔顿至700道尔顿。
在本发明的接合化合物中,考虑到化学性质、位阻和所连接的活性物质和肽或伪肽的数目,不同部分之间的偶联可通过任意可接受的结合方式进行。因此,偶联可在生理介质或在细胞内通过一或多个裂解或未裂解的共价键、离子键、氢键、疏水键或范德华键实现。此外,若需要经由S,D可在以下位点与V或P偶联:在各种反应基团(尤其是V的一或多个N末端和/或C末端)和/或在由构成V的天然或非天然氨基酸侧链所化学引入或携带的一或多个反应基团。
偶联可在肽或伪肽的任意位点进行,其中官能团(例如-OH、-SH、-CO2H、-NH2、-SO3H、-CN、-N3、-NCS、-PO2H、马来酰亚胺酯或琥珀酰亚胺酯)是天然存在的或已被引入。因此,感兴趣的治疗分子或诊断(或医学成像)试剂或任何其他分子(例如分子探针)可通过共价键,在N末端或C末端或在这个肽序列的天然或非天然氨基酸侧链所携带的反应基团上连接(偶联)至该肽或伪肽载体。
类似地,偶联可在活性物质或感兴趣物质(感兴趣的治疗分子、诊断或医学成像剂、任意其他分子,例如分子探针)的任意位点进行,其中,例如官能团(例如-OH、-SH、-CO2H、-NH2、-SO3H、-CN、-N3、-NCS、-PO2H、马来酰亚胺或琥珀酰亚胺酯)是天然存在的或已被引入。
这种偶联化学还适用于将肽或药物偶联至上述分子平台。
优选地,该相互作用是足够稳固的,使得所述肽在到达作用位点前无法脱离该活性物质。出于这一原因,本发明优选的偶联是共价偶联,但也可使用非共价偶联。可将感兴趣的物质在以下位点直接偶联至肽(直接连接合成):在一个末端(N端或C端)或在该序列的一种组成性氨基酸的侧链,或在所引入的任意官能团的多聚体。还可将感兴趣的物质在以下位点通过连接基团或间隔基偶联:在所述肽的一个末端,或在所述序列的一个组成性氨基酸的侧链(图1)。需要或不需要间隔基的共价化学偶联方式包括那些选自含有烷基、芳基、PEG或肽基团的双或多官能剂经酯、醛或烷基酸或芳基酸、酸酐、巯基或羧基、衍生自溴化氰或氯化氰、羰二咪唑、琥珀酰亚胺酯、磺酸卤化物、马来酰亚胺、叠氮化物、异硫氰酸酯、炔烃的基团的共价化学偶联。
在这一方面,本发明还涉及制备例如上文所定义的接合化合物的方法,其特征在于,该方法包括以下步骤:肽或伪肽或多聚体和物质D的偶联,若需要经由S,优选通过化学、生物化学或酶促途径,或通过基因工程。
本发明还涉及药物组合物,其特征在于,其包含至少一种例如上文所定义的接合化合物和一种或多种药学上可接受的赋形剂。
本发明还涉及诊断性组合物,其特征在于,其包含组成例如上文所定义的接合化合物的诊断或医学成像剂。
所述缀合物可以任意药学上可接受的盐的形式使用。表述“药学上可接受的盐”是指,例如且非限制性地为药学上可接受的碱或酸加成盐、水合物、酯、溶剂合物、前体、代谢物或立体异构体、所述载体或载有至少一种感兴趣物质的缀合物。
表述“药学上可接受的盐”是指无毒的盐,其通常可通过将游离碱与合适的有机或无机酸反应进行制备。这些盐保留了游离碱的生物学效应和性质。这些盐的代表性实例包括水溶性和水不溶性盐,例如乙酸盐、N-甲基葡糖胺铵盐、氨芪磺酸盐(4,4-二氨基芪-2,2’-二磺酸盐)、苯磺酸盐、苯甲酸盐、碳酸氢盐、硫酸氢盐、酒石酸氢盐、硼酸盐、氢溴酸盐、溴化物、丁酸盐(buryrates)、樟脑磺酸盐、碳酸盐、盐酸盐、氯化物、柠檬酸盐、克拉维酸盐、二氯水合物、二磷酸盐、依地酸盐、依地酸钙、乙二磺酸盐、丙酸酯十二烷基硫酸盐、乙磺酸盐、延胡索酸盐、葡庚糖酸盐、葡糖酸盐、谷氨酸盐、羟乙酰基对氨苯基砷酸盐(glycolylarsanylates)、六氟磷酸盐、己基间苯二酚酸盐(hexylresorcinates)、哈胺、羟基萘甲酸盐、碘化物、异硫代硫酸盐、乳酸盐、乳糖酸盐、月桂酸盐、苹果酸盐、马来酸盐、扁桃酸盐、甲磺酸盐、甲基溴化物、甲基硝酸盐、甲基硫酸盐、粘酸盐、萘磺酸盐、硝酸盐、3-羟基-2-萘甲酸盐、油酸盐、草酸盐、棕榈酸盐、双羟萘酸盐(1,1-亚甲基-双-2-羟基-3-萘甲酸酯或emboate)、泛酸盐、磷酸、苦味酸盐、聚半乳糖醛酸盐、丙酸盐、对甲苯磺酸盐、水杨酸盐、硬脂酸盐、碱式醋酸盐、琥珀酸盐、硫酸盐、磺基水杨酸盐、舒拉酸盐(suramates)、鞣酸盐、酒石酸盐、8-氯茶碱盐、甲苯磺酸盐、三乙基碘、三氟乙酸盐和戊酸盐。
本发明的组合物有利地包含药学上可接受的载体或赋形剂。所述药学上可接受的载体可根据各个给药方式选出通常所用的载体。根据所设定的给药方式,所述化合物可以是固体、半固体或液体形式。对于固体组合物,例如片剂、丸剂、粉剂,或分散的或包含在明胶胶囊中的颗粒剂,所述活性物质可与以下组合:a)稀释剂,例如乳糖、葡萄糖、蔗糖、甘露醇、山梨醇、纤维素和/或甘氨酸;b)润滑剂,例如二氧化硅、滑石、硬脂酸、其镁盐或钙盐和/或聚乙二醇;c)粘合剂,例如硅酸镁和硅酸铝、淀粉糊、明胶、黄蓍胶、甲基纤维素、羧甲基纤维素钠和/或聚乙烯吡咯烷酮;d)崩解剂,例如淀粉、琼脂、海藻酸或其钠盐,或泡腾合剂;和/或d)吸收剂、染料、调味剂和甜味剂。所述赋形剂可以是,例如,甘露醇、乳糖、淀粉、硬脂酸镁、糖精钠、滑石、纤维素、葡萄糖、蔗糖、碳酸镁和药物制品的类似物。对于半固体组合物,例如栓剂,所述赋形剂可以是,例如,乳液或油状混悬液或基于聚烷二醇,例如聚丙二醇。液体组合物,尤其是注射物或包含在软胶囊中的那些,可通过以下进行制备,例如,将活性物质溶解、分散等于药学纯的溶剂中,例如水、生理盐水溶液、葡萄糖水溶液、甘油、乙醇、油及其类似物。
可将本发明组合物或缀合物通过任意合适的途径进行给药,且非限制性地,例如,胃肠外给药途径,例如,胃肠外给药可皮下、静脉、肌内途径注射的剂型;口服途径(或经口),例如,口服给药包衣或未包衣片剂、明胶胶囊、粉剂、丹剂、混悬液或口服溶液剂型(该口服给药形式可以是立即释放剂型或缓释剂型或延长释放剂型);直肠途径,例如,直肠给药栓剂剂型;局部途径,尤其是经皮途径,例如,局部给药贴剂、香膏剂或凝胶剂型;鼻内途径,例如,鼻内给药气雾剂和喷雾剂剂型;经舌途径;或眼内途径。
该药物组合物通常包含有效量的本发明肽或伪肽或缀合物。本文所述的“治疗有效量”是指对于给定病症和给药方案,能提供治疗效果的剂量。通常,施用平均剂量的活性物质以明显改善与疾病或病理状态有关的一些症状。例如,在治疗脑癌或其他组织癌症、CNS病症、损伤或障碍时,降低、预防、延迟、消除或停止所述疾病或障碍的一个病因或症状的活性物质的剂量是治疗有效的。
“治疗有效量”的活性物质不一定会治愈疾病或障碍,但是能提供对这一疾病或障碍的治疗,使延迟、阻止或预防其表征,或减退其症状,或修改其症状或例如使症状没那么严重,或加速患者恢复。
应理解,个人的“治疗有效量”尤其取决于各种因素,包括活性物质的活性/效力、给药时间、给药途径、其消除速率及其代谢、药物组合/相互作用和以预防或治疗为主所治疗的疾病(或障碍)的严重性,以及患者的年龄、体重、总体健康状况、性别和/或饮食。
依据所偶联的物质,本发明的缀合物和组合物可用于治疗、预防、诊断或成像各种病症,尤其是影响CNS或眼的病症、感染性病症或癌症、炎性疾病和/或肝硬化或肝纤维化、肝癌、其他组织的疾病,例如肾上腺和肠。
在这个方面,本发明涉及上述药物缀合物或组合物在治疗或预防CNS病症或障碍、脑瘤或其他癌细胞和脑或其他组织的细菌、病毒、寄生虫或真菌感染性病症、炎性疾病和/或肝硬化或肝纤维化、肝癌、其他组织(例如肾上腺和肠)疾病中的用途。
本发明还涉及上述药物缀合物或组合物在诊断或成像CNS或眼病症或障碍、脑瘤或其他癌细胞和脑或其他组织的细菌、病毒、寄生虫或真菌感染性病症、炎性疾病和/或肝硬化或肝纤维化、肝癌、其他组织(例如肾上腺和肠)疾病中的用途。
本发明还涉及例如上文所定义的缀合物或组合物在治疗、成像和/或诊断脑瘤或其他癌细胞类型中的用途。研究已明确显示,患某些癌症的患者有低胆固醇血症。这个低胆固醇血症是癌细胞过度使用胆固醇的结果。残留的癌细胞会导致LDLR在肿瘤器官中表达水平升高(Henricksson等人,1989,Lancet,2(8673),1178-1180)。因此,LDLR在细胞与某些癌症中表达水平的升高之间存在联系。最近还显示,LDLR的数目在某些病理细胞(例如癌细胞)表面非常高。普遍认为,1000-3000个LDLR存在于非病理性细胞表面。类似地,LDLR很少存在于皮质灰质细胞(Pitas等人,1987,J.Biol.Chem.,262,14352-14360)。对于胶质母细胞瘤,已显示出LDLR的过表达。因此,在脑瘤细胞的表面,125,000(对于U-251细胞)-900,000(对于SF-767细胞)个LDLR已被计数(Malentiska等人,2000,CancerRes.,60,2300-2303;Nikanjam等人,2007,Int.J.Pharm.,328,86-94)。还应当注意的是,许多肿瘤细胞过度表达LDLR,例如以下癌症的细胞:前列腺癌(Chen等人,2001,Int.J.Cancer,91,41-45)、结肠癌(Niendorf等人,1995,Int.J.Cancer,61,461-464)、白血病(Tatidis等人,2002,Biochem.Pharmacol.,63,2169-2180)、结肠直肠癌(Caruso等人,2001,AnticancerRes.,21,429-433)、乳腺癌(Graziani等人,2002,Gynecol.Oncol.,85,493-497),以及肝癌、肾上腺癌、胰腺癌、卵巢癌、肺癌等。
本发明还涉及如上文所定义的缀合物或组合物在治疗、成像和/或诊断脑或其他组织的细菌、病毒、寄生虫或真菌感染病症中的用途,所述病症例如且非限制性地为AIDS或髓膜炎等。LDLR还存在于肝细胞。目前已知的,丙型肝炎病毒(HCV)的胞吞作用可经由LDLR发生。LDLR可在人肝细胞感染HCV早期作为病毒受体(Molina等人,2007,J.Hepatol.,46(3),411-419)。因此本发明缀合物可用于特异性地靶向被病毒(例如表达LDLR和/或经由LDLR来调节健康细胞的病毒感染过程的乙型肝炎和丙型肝炎病毒)感染的病理细胞。本发明还涉及如上文所定义的缀合物或组合物在治疗、成像和/或诊断其他肝病(例如且非限制性地,饮酒所导致的病毒性肝炎或非饮酒所导致的病毒性肝炎、急性肝炎、肝硬化或肝纤维化、原发性胆汁性肝硬化和肝胆道的其他疾病等)中的用途(Poeltra等人,2012,JControlRelease,161(2):188-97)。
本发明还涉及如上文所定义的缀合物或组合物在成像或治疗富集LDLR的肾上腺或肠病中的用途。
本发明还涉及如上文所定义的缀合物或组合物在治疗、成像和/或诊断神经退行性病症(例如,非限制性地,阿尔茨海默病、帕金森病、克雅病、脑血管意外(CVA)、牛海绵状脑病、多发性硬化、肌萎缩侧索硬化、脊髓损伤等)中的用途。
本发明还涉及如上文所定义的缀合物或组合物在治疗、成像和/或诊断神经疾病(例如,非限制性地,癫痫、偏头痛、脑炎、CNS疼痛等)中的用途。
本发明还涉及如上文所定义的缀合物或组合物在治疗、成像和/或诊断神经变性和兴奋性毒素过程(例如,非限制性地,瞬时心脏呼吸停止、中风、新生儿缺血等的结果)中的用途。
本发明还涉及如上文所定义的缀合物或组合物在治疗、成像和/或诊断神经精神疾病(例如,非限制性地,抑郁症、孤独症、焦虑症、精神分裂症等)中的用途。
本发明还涉及如上文所定义的缀合物或组合物在治疗、成像和/或诊断眼病(例如,非限制性地,色素性视网膜炎、黄斑变性、视锥-视杆细胞营养不良(CORD)、视网膜脱离、视网膜剥落、高血压性视网膜病变和糖尿病视网膜病变、视网膜母细胞瘤、视网膜脂血症等)中的用途。
术语“治疗”和其他类似表述是指获得药理学和/或生理学效果,例如,抑制癌细胞生长、癌细胞死亡或改善疾病或神经或眼部疾病。该效果可以是预防性的,以完全或部分预防患者疾病或该疾病的症状的恶化或预防其在健康受试者中的传播,和/或可以是治疗性的,以完全或部分治疗疾病和/或其相关有害影响。本文所用术语“治疗”涵盖了对哺乳动物,尤其是人的任意疾病治疗,且包括:(a)预防疾病(例如,预防癌症)或病症,其可在易感这一病症或障碍的人中出现,但其还尚未确诊患有该病症或障碍,(b)减缓疾病(例如,停止其发展),或(c)缓解疾病(例如,减少疾病相关症状)。该术语“治疗”还包括活性物质的任意施用,以管理、治愈、缓解、改善、降低或抑制个体或患者的症状,包括,但不限于,施用有此需要的个人由本文所述的组成载体或缀合物的药物。
本发明还涉及本发明的肽或伪肽在提高其偶联的活性物质或感兴趣的物质(感兴趣的治疗分子、诊断或医学成像剂或任何其他分子,例如分子探针)的生物活性中的用途。
本发明还涉及本发明的肽或伪肽在降低其偶联的活性物质或感兴趣的物质(感兴趣的治疗分子、诊断或医学成像剂或任何其他分子,例如分子探针)的毒性中的用途。
在了解了以下实施例后,本发明的其他方面和优势将会变得明显,以下实施例仅是示例性的且并不限制本申请的范围。
实施例
实施例I
肽的合成以及与示踪分子(生物素、荧光素、若丹明、青色素或具有酶活性的Stag肽)的偶联。
肽是通过固相肽合成(SPPS)方法合成的,其在AdvancedChemTechApex396(AAPPTec)合成器或LibertyTM(CEM)微波合成器上,使用Fmoc/tBu策略在聚苯乙烯-1%DVB上的RinkAmideAM树脂、在聚苯乙烯-1%DVB上的Wang、在聚苯乙烯-1%DVB上的Barlos(2-氯三苯甲基氯)或在聚苯乙烯-1%DVB上的SieberAmide。依据所使用的树脂,该负载(或取代)为0.25mmol/g至1.6mmol/g。
经Fmoc进行N保护的氨基酸(或者一些N末端是经Boc所保护)和/或在它们的侧链被垂直官能团(尤其是对酸敏感的官能团)所保护的氨基酸、化学偶联和脱保护试剂以及溶剂,均从专业公司购入并以原样使用。
RinkAmide和Wang树脂使能够合成侧链及其C末端完全脱保护的肽序列。因此,这是二维(Fmoc/tBu)正交SPPS。
Barlos和Sieber高度对酸敏感(HAL)树脂分别使末端(C端)酸或酰胺官能团释放,同时保留了对合成肽的各个氨基酸垂直侧的保护以及对最后一个氨基酸的胺官能团末端(N端)胺的保护(例如,针对新合成的肽序列稳定性的问题,进行N-乙酰化保护)。经由Fmoc(Prot1)合成策略的这些树脂类型,使得能够使用对酸敏感的垂直侧的保护(Prot2:Boc、tBu、OtBu、Trt、Mmt、Acm等),其仅在强酸介质中裂解,而所保护的肽在非常弱的酸性条件下裂解。这种裂解类型使得能够复原其侧链官能团完全被保护(Prot2)的肽序列,尤其是用以将感兴趣的治疗分子与肽偶联。因此,这是三维(Barlos或Sieber/Fmoc/tBu)正交SPPS。
在肽合成过程中,针对各个氨基酸所使用的标准垂直侧的保护(Prot2)为:Arg(N-Pbf)、Arg(N-Pmc)、Asn(N-Trt)、Asp(O-tBu)、Cys(S-Acm)、Cys(S-Mmt)、Cys(S-4MeBn)、Cys(S-tBu)、Cys(S-Tmob)、Cys(S-Trt)、Glu(O-tBu)、Gln(N-Trt)、His(N-Trt)、Lys(N-Boc)、Pen(S-Acm)、Pen(S-Trt)、Ser(O-tBu)、Thr(O-tBu)、Trp(N-Boc)、Tyr(O-tBu)(AppliedBiosystems,1998,Cleavage,Deprotection,和IsolationofPeptidesafterFmocSynthesis.TechnicalBulletin)。Gly、Sar、Ala、Val、Leu、Ile、Phe、Met、Pro、Pip和Thz没有侧保护,因为他们各自的化学结构不需要侧保护。
氨基酸是通过以下偶联的:使用DIEA/HBTU/HOBt或DIPC/HOBt的DMF溶液来活化n+1氨基酸的酸性官能团。
因此偶联的新氨基酸的Fmoc(Prot1)基团的脱保护是使用20%哌啶的DMF溶液进行的。
在肽测序过程中,最后一个偶联的氨基酸可以被Boc官能团保护(以在合成结束时释放其自由末端胺官能团),或被乙酰化(例如以稳定所合成的新肽,并且在共价偶联感兴趣的治疗分子至C端位置的过程中,降低副反应的风险)或被丙酰化。
根据所合成的肽,二硫键是由两个被适当保护的Cys(Acm、Trt、tBu等)的两个硫醇官能团的分子内环化得到的,无论是在溶液或是在树脂上,使用本领域技术人员常用的试剂:H2O/AcOH/,/(NH4)2CO3/DMSO、H2O/AcOH,/(NH4)2CO3、I2/DMF、I2/HF
用于药物递送的组合物和方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












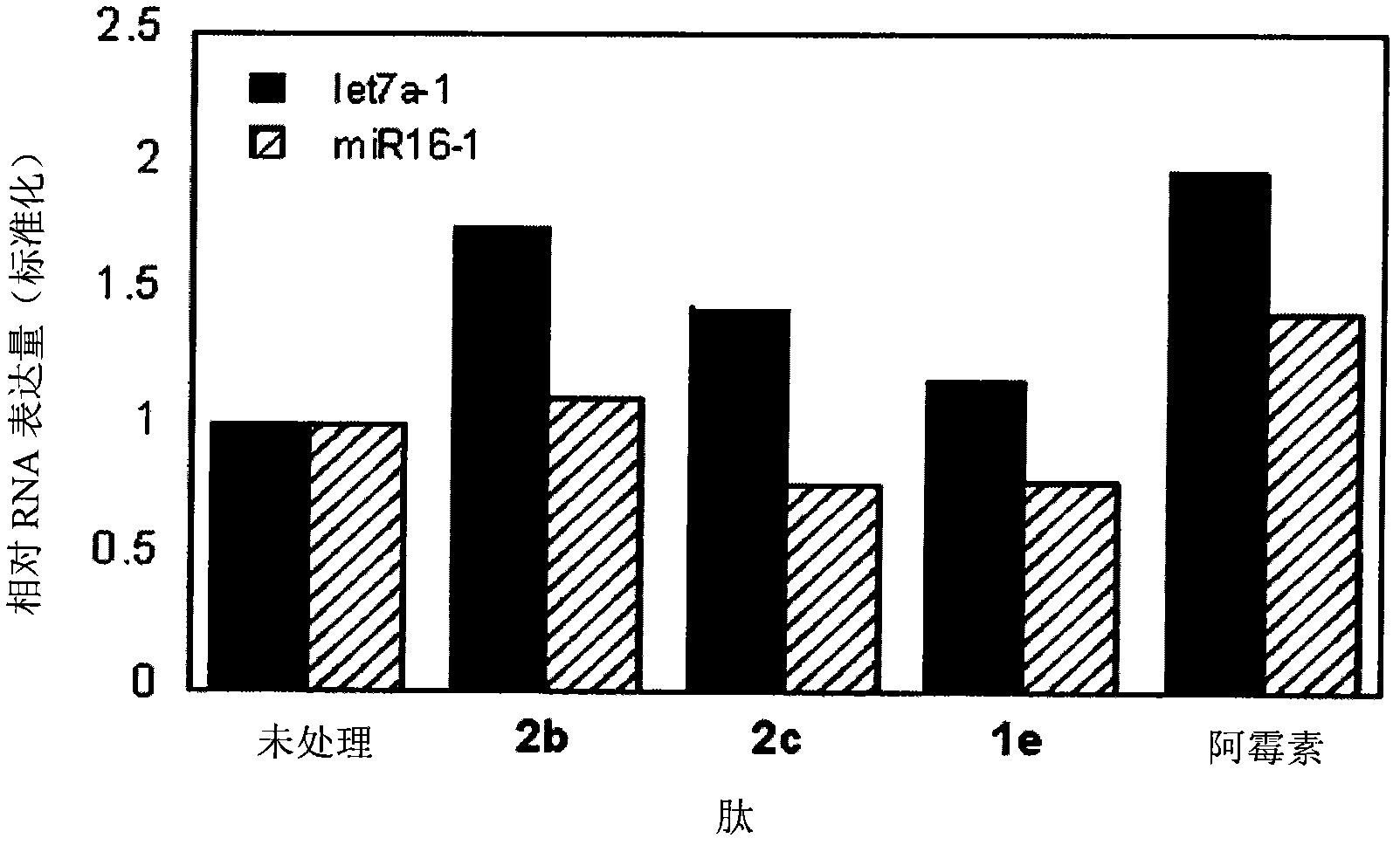




动态评分
0.0