

IPC分类号 : C07F3/06I,C07F5/00I,C07F11/00I,C07F7/00I,C07F1/10I,C07F13/00I,C09K9/02I







专利摘要
本发明公开了一种合成光致变色晶态无机有机杂化配合物的方法,将金属源、有机配体和DESs的混合物在100~160℃密封条件下反应2~15天,所述金属源为含有锆、钨、锰、锌、铕、银和镓的金属源中的一种,所述DESs的组分中含有氯化胆碱,所述金属源、有机配体和DESs中氯化胆碱的摩尔比为:0.5~2:1~3:5~20。本发明通过离子热法合成的新型晶态无机有机杂化的光致变色材料,兼具有机光致变色材料和无机光致变色材料的优势,具有成本低、环境友好的优点。
权利要求
1.一种合成光致变色晶态无机有机杂化配合物的方法,其特征在于:
将金属源、有机配体和DESs的混合物在100~160℃密封条件下反应2~15天,所述金属源为含有锆、钨、锰、锌、铕、银和镓的金属源中的一种,所述DESs的组分中含有氯化胆碱,所述金属源、有机配体和DESs中氯化胆碱的摩尔比为:0.5~2:1~3:5~20,其中,所述有机配体为4,4'-联吡啶或2,2'-联吡啶。
2.如权利要求1所述的方法,其特征在于:所述DESs为氯化胆碱和草酸、氯化胆碱和丙二酸、氯化胆碱和苯二甲酸、氯化胆碱和二甘醇酸、氯化胆碱和柠檬酸或氯化胆碱和乙二醇中的一种。
3.如权利要求2所述的方法,其特征在于:所述DESs中氯化胆碱与另一组分的的摩尔比为1:1~4。
4.如权利要求1所述的方法,其特征在于:其中反应物中还包括酸。
5.如权利要求4所述的方法,其特征在于:所述酸为磷酸、亚磷酸或氢氟酸。
6.如权利要求4所述的方法,其特征在于:所述金属源、有机配体、酸和DESs中氯化胆碱的摩尔比为:0.5~2:1~3:0~10:5~20。
说明书
技术领域
本发明涉及光致变色材料技术领域,具体涉及一种合成光致变色晶态无机有机杂化配合物的方法。
背景技术
光致变色行为是指某些材料在一定强度和波长的光照作用下发生颜色变化且在加热或光照条件下可实现颜色可逆恢复的现象,两种状态的相互转换通常伴随着某些物理化学性质的变化,如氧化还原电位、电导率、荧光、光电流、光电压等等。近年来,光致变色材料因在信息存储元件、装饰和防护包装材料、自显影全息记录照相、防伪、国防等领域内存在的实际或潜在的应用价值,尤其是在晶态光致变色材料的研究领域,引起了科研工作者的广泛关注。其中,有机光致变色材料(如二芳基乙烯类,螺吡喃、螺恶嗪、俘精酸酐、偶氮化合物等)已经被广泛研究,它们光响应速度快,颜色丰富多样且可加工性好,但大部分不耐高温且容易疲劳失活;无机光致变色材料(如过渡金属氧化物、金属卤化物、多金属氧酸盐等)虽通常具有较好的光热稳定性和耐疲劳性,但褪色困难、消色慢,且相比有机光致变色材料而言种类稀少。光致变色材料根据加工需求需要制备成不同的聚集态(如膜,纳米纤维,镀层,晶片等),这使得光致变色材料能够兼具优良的光致变色性能和高的光热稳定性及可加工性,单纯的无机或有机光致变色材料均难满足这一要求。因此,开发能够兼具无机光致变色材料和有机光致变色材料优点的新型无机有机杂化光致变色材料已成为研究热点。
近年来,将有机光致变色分子作为模板剂客体分子或配体分子引入到无机有机杂化配位聚合物材料中以制备晶态无机有机光致变色材料引起了人们越来越多的关注,并已取得了一些研究成果。福建物质结构研究所的郭国聪课题组、张杰课题组,吉林大学于吉红课题组,郑州大学臧双全课题组,华东师范大学高恩庆课题组,华南理工大学傅志勇课题组等都在这一领域开展了一系列的工作。迄今为止,晶态无机有机杂化光致变色材料多是基于紫精(MV)衍生物、萘二酰亚胺(NDI)衍生物、二芳基乙烯衍生物为配体的金属有机骨架材料(MOFs),其配体往往需要精细的设计,其复杂的合成及性能表现使之仍远未被市场接受。在新型晶态无机有机杂化光致变色材料的合成中,寻求最大限度地有效利用源化学品,并避免使用有毒和挥发性的有机溶剂的反应途径具有巨大的优势和重要的意义。
发明内容
本发明的目的是提供一种兼具无机光致变色材料和有机光致变色材料优点、且环境友好的光致变色晶态无机有机杂化配合物的合成方法。
为实现上述目的,本发明采用的技术方案为:
一种合成光致变色晶态无机有机杂化配合物的方法,具体如下:
将金属源、有机配体和DESs的混合物在100~160℃密封条件下反应2~15天,所述金属源为含有锆、钨、锰、锌、铕、银和镓的金属源中的一种,所述DESs的组分中含有氯化胆碱,所述金属源、有机配体和DESs中氯化胆碱的摩尔比为:0.5~2:1~3:5~20。
进一步地,所述有机配体为4,4'-联吡啶或2,2'-联吡啶。
进一步地,所述DESs为氯化胆碱和草酸、氯化胆碱和丙二酸、氯化胆碱和苯二甲酸、氯化胆碱和二甘醇酸、氯化胆碱和柠檬酸或氯化胆碱和乙二醇中的一种。
进一步地,所述DESs中氯化胆碱与另一组分的的摩尔比为1:1~4。
进一步地,其中反应物中还包括酸。
进一步地,所述酸为磷酸、亚磷酸或氢氟酸。
进一步地,所述金属源、有机配体、酸和DESs中氯化胆碱的摩尔比为:0.5~2:1~3:0~10:5~20。
本发明提供的一种合成光致变色晶态无机有机杂化配合物的方法,通过离子热法合成新型晶态无机有机杂化光致变色材料,兼具有机光致变色材料和无机光致变色材料的优势,具有成本低、环境友好的优点。
附图说明
图1为本发明实施例1提供的所得晶体的扫描电子显微镜图。
图2为本发明实施例1提供的所得晶体的变色前后和热褪色后的粉末X射线衍射图谱。
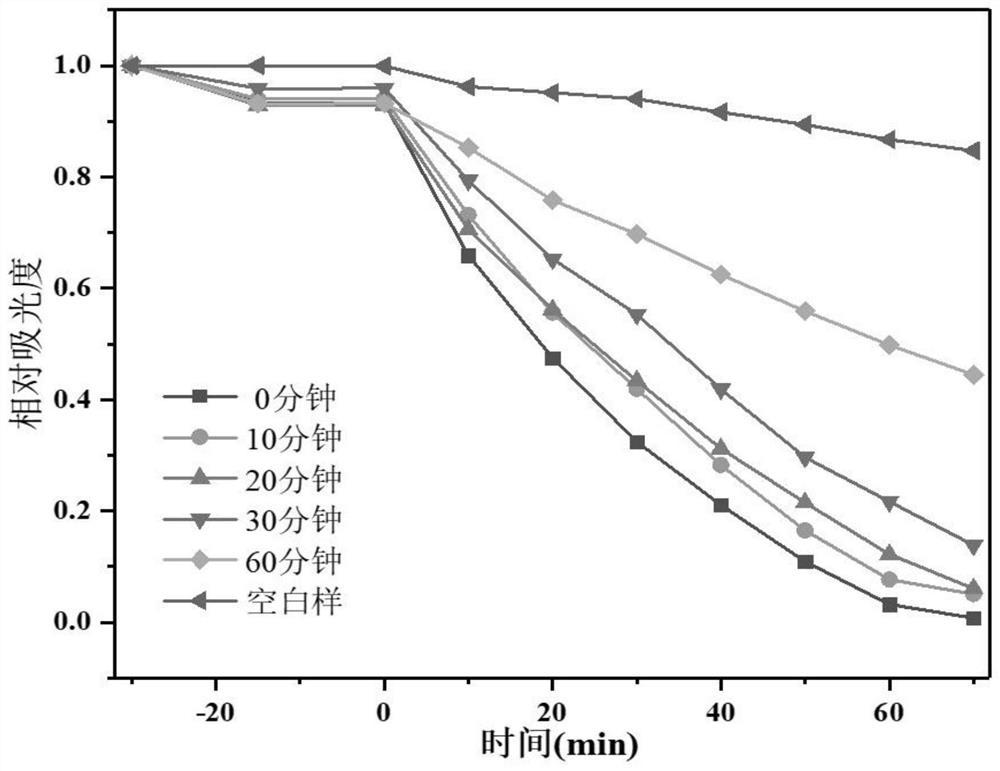
图3为本发明实施例1提供的所得晶体的固体紫外可见吸收光谱图。
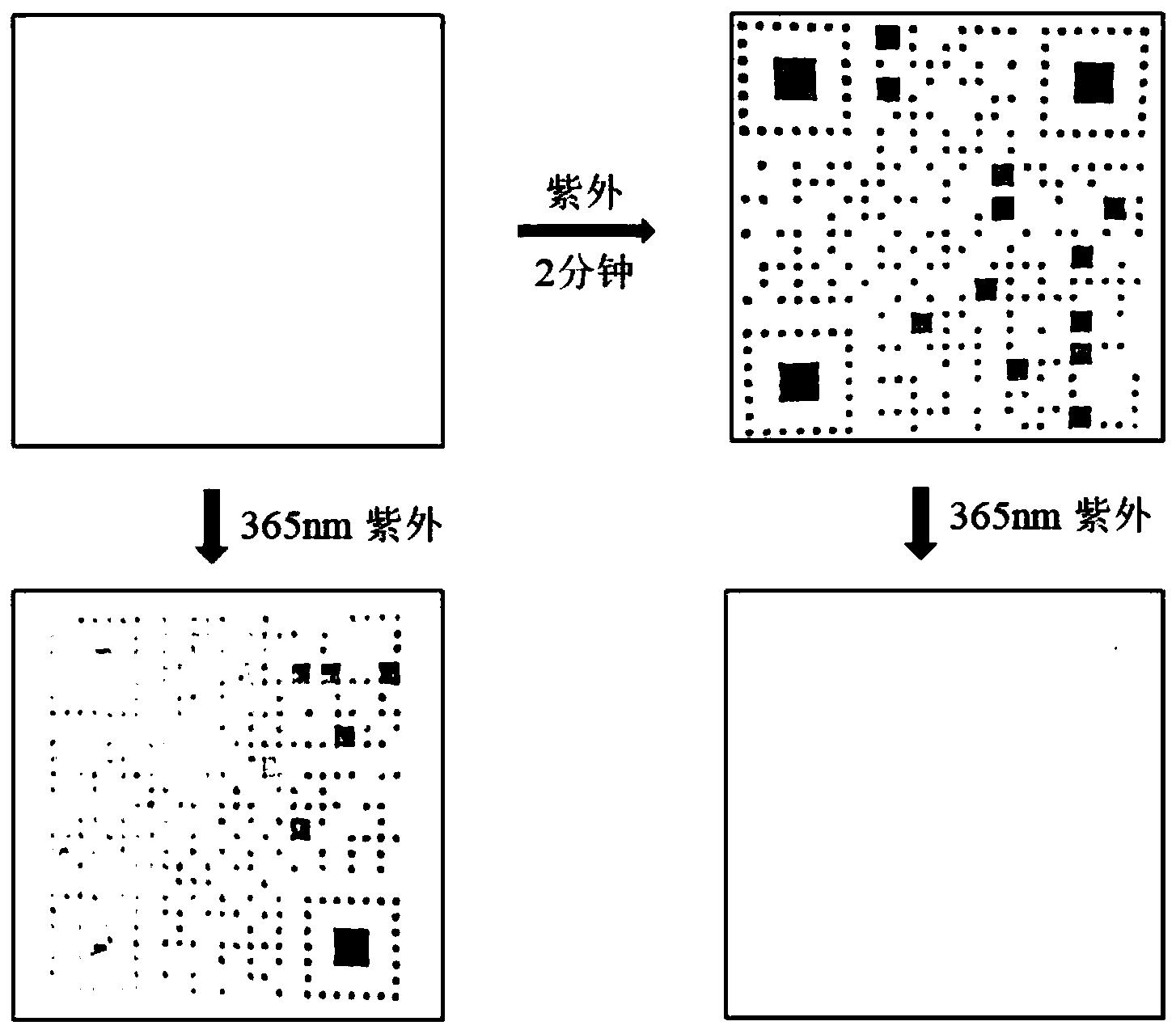
图4为本发明实施例1提供的所得晶体制得的滤纸膜的光致变色行为效果图。
图5为本发明实施例2提供的所得晶体的扫描电子显微镜图。
图6为本发明实施例2提供的所得晶体的变色前后和热褪色后的粉末X射线衍射图谱。
图7为本发明实施例2提供的所得晶体的固体紫外可见吸收光谱图。
图8为本发明实施例2提供的所得晶体的光致变色行为效果图。
图9为本发明实施例3提供的所得晶体的变色前后和热褪色后的粉末X射线衍射图谱。
图10为本发明实施例4提供的所得晶体的变色前后和热褪色后的粉末X射线衍射图谱。
图11为本发明实施例5提供的所得晶体的变色前后和热褪色后的粉末X射线衍射图谱。
图12为本发明实施例6提供的所得晶体的变色前后和热褪色后的粉末X射线衍射图谱。
具体实施方式
本发明实施例提供一种合成光致变色晶态无机有机杂化配合物的方法,具体如下:将一定量的金属源、有机配体和DESs的混合物在100~160℃密封条件下反应2~15天。待反应完成后于室温下自然冷却,将产物移出后用去离子水反复超声洗涤3~7次,40~80℃温度条件下烘干,最后得到所需晶体。
其中,所述金属源为含有锆、钨、锰、锌、铕、银和镓的金属源中的一种;所述有机配体为4,4'-联吡啶或2,2'-联吡啶;所述DESs为氯化胆碱和草酸、氯化胆碱和丙二酸、氯化胆碱和苯二甲酸、氯化胆碱和二甘醇酸、氯化胆碱和柠檬酸或氯化胆碱和乙二醇中的一种;所述DESs中氯化胆碱与另一组分的的摩尔比为1:1~4;所述金属源、有机配体和DESs中氯化胆碱的摩尔比为:0.5~2:1~3:5~20。
其中,反应物中还包括酸;所述酸为磷酸、亚磷酸或氢氟酸;所述金属源、有机配体、酸和DESs中氯化胆碱的摩尔比为:0.5~2:1~3:0~10:5~20。
有机配体联吡啶由于其自身特殊的电子缺陷性质有与富电子物质形成电荷转移络合物的倾向,在某些情况下具有通过化学、电学或光刺激进行电子转移的氧化还原能力,故本发明选用联吡啶作为有机配体。同时,我们选择低共熔混合物(DESs)这一新的离子液体体系,因组成DESs的组分通常会通过氢键相互作用自缔合而使混合物熔点低于每种单独组分的熔点,使合成过程可在常压和较低的温度下进行反应,从而避免了传统合成中高温高压所带来的安全隐患,适应了当前所倡导的清洁技术和可持续发展的要求。即:本发明选用廉价易得的金属源(连接点)和非光致变色有机配体(电子供体和受体),在组成多样且廉价易得的DESs体系中,采用离子热合成法合成了一系列变色材料。反应过程中,DESs组成中的离子盐会分解解离出氢基或烷基基团,进而与有机分子重组。合成的光致变色晶态无机有机杂化配合物兼具有机光致变色材料和无机光致变色材料的优势,具有光响应速度快、颜色丰富多样、可加工性好、较好的光热稳定性和耐疲劳性、褪色迅速,种类丰富的特点。此外,合成过程配比较简单,产率较高且易于放大合成。
在光致变色晶体材料的合成中,可以选择常用的磷酸和亚磷酸来调控体系酸度,或通过可促进晶体晶化的矿化剂氢氟酸来调控体系酸度。通过大量工艺参数的调整实验,最终确定反应温度应控制在100~160℃,反应时间为2~15天,金属源、有机配体、酸和DESs中氯化胆碱摩尔比范围为:0.5~2:1~3:0~10:5~20。可通过调控金属源、有机配体、体系酸度、DESs组成和用量、反应温度和时间来调控产物的骨架组成和配位方式,制备出一系列具有不同结构及骨架组成的晶态无机有机杂化光致变色材料,为低成本大规模商业化合成功能性光致变色材料提供了一条有效的合成途径。
实施例1一种合成光致变色晶态无机有机杂化配合物的方法
以硝酸锌、4,4’-联吡啶、亚磷酸、氯化胆碱和草酸作为离子溶剂,在100℃温度下反应7天,合成新型晶态无机有机杂化配合物。
具体如下:将硝酸锌1mmol、4,4’-联吡啶1mmol、亚磷酸4mmol、氯化胆碱11mmol、草酸20mmol加入到25mL聚四氟乙烯为衬里的不锈钢釜中,然后将反应釜密封后放入100℃的烘箱内加热7天。待反应完成后将反应釜取出放于室温下自然冷却,将产物移出,用去离子水反复超声洗涤3次,70℃温度条件下烘干。
合成产物表征情况如下:如图1所示,所得晶体为条形,粒径约为100×40μm。如图2所示,所得晶体原样、变色及加热褪色后的样品的粉末XRD谱图对应得很好,说明样品结构稳定。如图3所示,所得晶体随紫外光照时间变化的固体紫外吸收光谱表明该配合物在385nm、554nm和620nm处有吸收峰,这些吸收峰对应于质子化的H2bpy
实施例2一种合成光致变色晶态无机有机杂化配合物的方法
以硫酸锰、4,4’-联吡啶、亚磷酸、氯化胆碱和二甘醇酸作为离子溶剂,在140℃温度下反应3天,合成新型晶态无机有机杂化配合物。
将硫酸锰1mmol、4,4’-联吡啶2mmol、亚磷酸1mmol、氯化胆碱5mmol、二甘醇酸20mmol加入到25mL聚四氟乙烯为衬里的不锈钢釜中,然后将反应釜密封后放入140℃的烘箱内加热3天。待反应完成后将反应釜取出放于室温下自然冷却,将产物移出,用去离子水反复超声洗涤3次,80℃温度条件下烘干。
合成产物表征情况如下:如图5所示,所得晶体为条形,粒径约为75×25μm。如图6所示,所得晶体原样、变色及加热褪色后的样品的XRD谱图对应得很好,说明样品结构稳定。如图7所示,所得晶体随紫外光照时间变化的固体紫外吸收光谱表明该配合物在385nm、554nm和620nm处有吸收峰,这些吸收峰对应于质子化的H2bpy
实施例3一种合成光致变色晶态无机有机杂化配合物的方法
以硝酸银、4,4’-联吡啶、磷酸、氯化胆碱和丙二酸作为离子溶剂,在120℃温度下反应15天,合成新型晶态无机有机杂化配合物。
将硝酸银0.5mmol、4,4’-联吡啶3mmol、磷酸10mmol、氯化胆碱5.5mmol、丙二酸6mmol加入到25mL聚四氟乙烯为衬里的不锈钢釜中,最后将反应釜密封后放入120℃的烘箱内加热15天。待反应完成后将反应釜取出放于室温下自然冷却;将产物移出,用去离子水反复超声洗涤3次,70℃温度条件下烘干。
合成产物表征情况如下:如图9所示,所得晶体原样、变色及加热褪色后的样品的XRD谱图对应得很好,说明样品结构稳定。将所得晶体在48W的紫外灯照射下可迅速变为紫色,140℃加热30min可实现褪色,是一种新型晶态无机有机杂化光致变色材料。
实施例4一种合成光致变色晶态无机有机杂化配合物的方法
以六水合硝酸铕、2,2’-联吡啶、亚磷酸、氯化胆碱和草酸作为离子溶剂,在120℃温度下反应7天,合成新型晶态无机有机杂化配合物。
将六水合硝酸铕1mmol、2,2’-联吡啶1mmol、亚磷酸4mmol、氯化胆碱5.5mmol、草酸10mmol加入到25mL聚四氟乙烯为衬里的不锈钢釜中,最后将反应釜密封后放入120℃的烘箱内加热7天。待反应完成后将反应釜取出放于室温下自然冷却;将产物移出,用去离子水反复超声洗涤3次,70℃温度条件下烘干。
合成产物表征情况如下:如图10所示,所得晶体原样、变色及加热褪色后的样品的XRD谱图对应得很好,说明样品结构稳定。所得晶体在48W的紫外灯照射下,可迅速变为紫色,140℃加热30min可实现褪色,是一种新型晶态无机有机杂化光致变色材料。
实施例5一种合成光致变色晶态无机有机杂化配合物的方法
以氧化镓、4,4’-联吡啶、氯化胆碱和草酸作为离子溶剂,在140℃温度下反应11天,合成新型晶态无机有机杂化配合物。
将氧化镓0.5mmol、4,4’-联吡啶1mmol、氯化胆碱10mmol、草酸20mmol加入到25mL聚四氟乙烯为衬里的不锈钢釜中,最后将反应釜密封后放入140℃的烘箱内加热11天。待反应完成后将反应釜取出放于室温下自然冷却;将产物移出,用去离子水反复超声洗涤3次,70℃温度条件下烘干。
合成产物表征情况如下:如图11所示,所得晶体原样、变色及加热褪色后的样品的XRD谱图对应得很好,说明样品结构稳定。所得晶体材料在48W的紫外灯照射下,所得晶体可迅速变为紫色,140℃加热30min可实现褪色,是一种新型晶态无机有机杂化光致变色材料。
实施例6一种合成光致变色晶态无机有机杂化配合物的方法
以氧化镓、4,4’-联吡啶、氢氟酸、氯化胆碱和乙二醇作为离子溶剂,在160℃温度下反应3天,合成新型晶态无机有机杂化配合物。
将氧化镓1mmol、4,4’-联吡啶1.5mmol、氢氟酸5.18mmol、氯化胆碱10mmol、乙二醇12mmol加入到25mL聚四氟乙烯为衬里的不锈钢釜中,最后将反应釜密封后放入160℃的烘箱内加热3天。待反应完成后将反应釜取出放于室温下自然冷却;将产物移出,用去离子水反复超声洗涤3次,70℃温度条件下烘干。
合成产物表征情况如下:如图12所示,所得晶体原样、变色及加热褪色后的样品的XRD谱图对应得很好,说明样品结构稳定。所得晶体材料在48W的紫外灯照射下,所得晶体可迅速变为紫色,140℃加热30min可实现褪色,是一种新型晶态无机有机杂化光致变色材料。
实施例7一种合成光致变色晶态无机有机杂化配合物的方法
以四氯化锆、4,4’-联吡啶、磷酸、氯化胆碱和苯二甲酸作为离子溶剂,在150℃温度下反应3天,合成新型晶态无机有机杂化配合物。
将四氯化锆1mmol、4,4’-联吡啶1.0mmol、亚磷酸0.5mmol、氯化胆碱20mmol、苯二甲酸24mmol加入到25mL聚四氟乙烯为衬里的不锈钢釜中,最后将反应釜密封后放入150℃的烘箱内加热3天。待反应完成后将反应釜取出放于室温下自然冷却;将产物移出,用去离子水反复超声洗涤3次,70℃温度条件下烘干。
合成产物表征情况如下:所得晶体原样、变色及加热褪色后的样品的XRD谱图对应得很好,说明样品结构稳定。所得晶体材料在48W的紫外灯照射下,所得晶体可迅速变为紫色,140℃加热30min可实现褪色,是一种新型晶态无机有机杂化光致变色材料。
实施例8一种合成光致变色晶态无机有机杂化配合物的方法
以钨酸钠、2,2’-联吡啶、亚磷酸、氯化胆碱和柠檬酸作为离子溶剂,在120℃下反应2天,合成新型晶态无机有机杂化配合物。
将钨酸钠2mmol、4,4’-联吡啶1mmol、亚磷酸0.5mmol、氯化胆碱5mmol、柠檬酸12.5mmol加入到25mL聚四氟乙烯为衬里的不锈钢釜中,最后将反应釜密封后放入120℃的烘箱内加热2天。待反应完成后将反应釜取出放于室温下自然冷却;将产物移出,用去离子水反复超声洗涤3次,70℃温度条件下烘干。
合成产物表征情况如下:所得晶体原样、变色及加热褪色后的样品的XRD谱图对应得很好,说明样品结构稳定。所得晶体材料在48W的紫外灯照射下,所得晶体可迅速变为紫色,140℃加热30min可实现褪色,是一种新型晶态无机有机杂化光致变色材料。
实施例9一种合成光致变色晶态无机有机杂化配合物的方法
以氧化镓、4,4’-联吡啶、氢氟酸、氯化胆碱和草酸作为离子溶剂,在140℃温度下反应3天,合成新型晶态无机有机杂化配合物。
将氧化镓5mmol、4,4’-联吡啶7.5mmol、氢氟酸25.9mmol、氯化胆碱50mmol、草酸60mmol加入到100mL聚四氟乙烯为衬里的不锈钢釜中,最后将反应釜密封后放入140℃的烘箱内加热3天。待反应完成后将反应釜取出放于室温下自然冷却;将产物移出,用去离子水反复超声洗涤3次,70℃温度条件下烘干。
合成产物表征情况如下:所得晶体材料在48W的紫外灯照射下,所得晶体可迅速变为紫色,140℃加热30min可实现褪色,是一种新型晶态无机有机杂化光致变色材料。经粉末X射线衍射分析,放大五倍所合成的晶体与之前所得晶体的XRD谱图保持一致,说明此种方法可应用于放大合成,更便于应用于工业大规模生产。
最后所应说明的是,以上具体实施方式仅用以说明本发明的技术方案而非限制,尽管参照实例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
一种合成光致变色晶态无机有机杂化配合物的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0