









专利摘要
一种含酸油催化脱酸方法,包括将含酸烃油原料引入反应器,先与保护剂接触,脱除原料油中的残碳前驱物,然后与催化脱酸催化剂接触进行脱酸反应得到酸值降低的烃油;所述的保护剂床层温度为200~380℃,所述的催化剂床层温度为200~380℃,反应器中水和二氧化碳的合并分压不大于350kPa、重时空速为0.1~50hr-1。本发明提供的含酸油催化脱酸方法,在脱酸催化剂前设置大孔径的保护剂,保护剂可以容纳较多的残炭前驱物,减少了这部分物质在脱酸催化剂上的吸附,使得脱酸催化剂具有较高的脱酸活性和稳定性。
说明书
技术领域技术领域
本发明涉及一种脱除原油及馏分油中石油酸的方法。
技术背景背景技术
随着石油资源的日益枯竭和采油技术不断提高,原油的开采范围不断扩大,高酸值原油的产量逐年增加。原油酸值过高会对炼油设备造成严重的腐蚀,影响炼油装置的长周期安全运转,增加加工成本,并且导致石油产品的酸值高,影响油品的使用。酸值(TAN)是中和1g原油中各种酸性组分所消耗的KOH的总量,以mgKOH/g表示(参见ASTMD-664方法)。通常的酸值大于0.5mgKOH/g的原油为含酸原油,酸值大于1.0mgKOH/g的原油为高酸原油。
原油中的酸包括环烷酸、脂肪酸、芳香酸、无机酸、硫醇、硫化氢和苯酚等,其中环烷酸的含量约占90%,对加工设备的腐蚀主要是由环烷酸引起。目前,含酸原油及馏分油脱环烷酸的生产技术主要包括:化学萃取法、吸附分离法、溶剂抽提法、酯化脱酸法、催化加氢脱酸法、热脱酸、催化热解脱酸法等。催化热解脱酸是在油品中加入催化剂,促进石油酸脱羧,可在较低的反应温度下反应,且脱酸率高。
CN1272869A中公开了在没有氢气存在的条件下,用第VB、VIB、VIIB、VIII族金属的油溶性或油分散性金属化合物的催化剂在204~426℃,大气压15~1000psi,维持水和CO2合并分压低于50psi的条件下对原油或馏分油进行催化脱羧。
US2006016723A1中公开了用碱土金属氧化物、过渡金属氧化物和稀土金属氧化物以及高岭土等粘土吸附剂相结合对原油进行催化脱羧反应,反应温度为200~450℃,反应系统可以是密封玻璃管、高压釜、流动反应器、间歇反应器、浆液反应器以及它们的组合等。
CN 1903991A公布了一种烃原料催化脱酸方法,包括将所述的烃原料在100~300℃与脱酸催化剂接触反应,所述的脱酸催化剂包括氧化钙和硫酸钙,其中氧化钙与硫酸钙的质量比为0.1~2.0。
但是现有催化热脱酸方法所用催化剂脱酸活性降低很快,催化剂寿命都相对比较短。
发明内容发明内容
本发明要解决的问题是现有技术中含酸油脱酸催化剂活性降低快,寿命短,提供一种催化剂活性高、寿命长的含酸油催化脱酸方法。
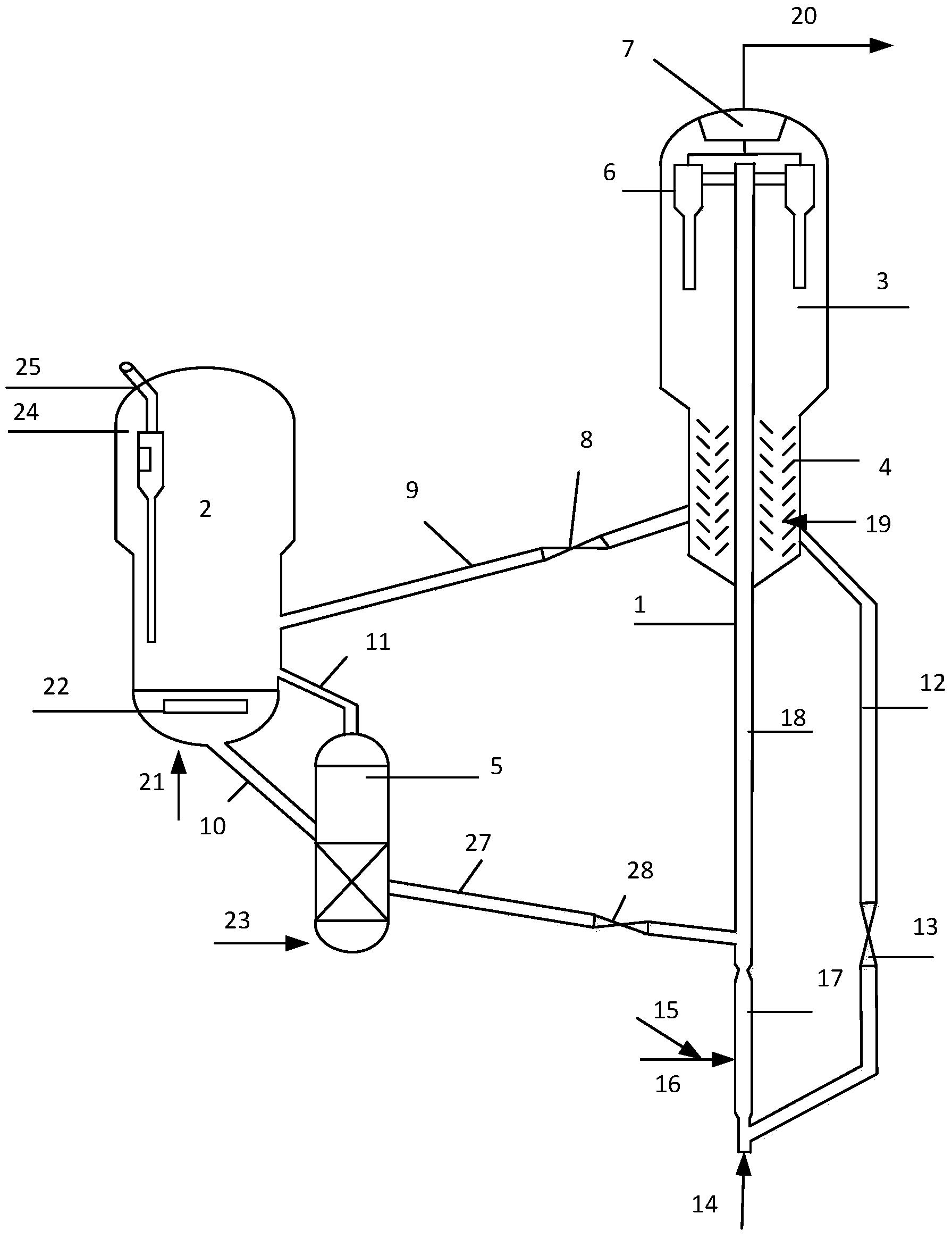
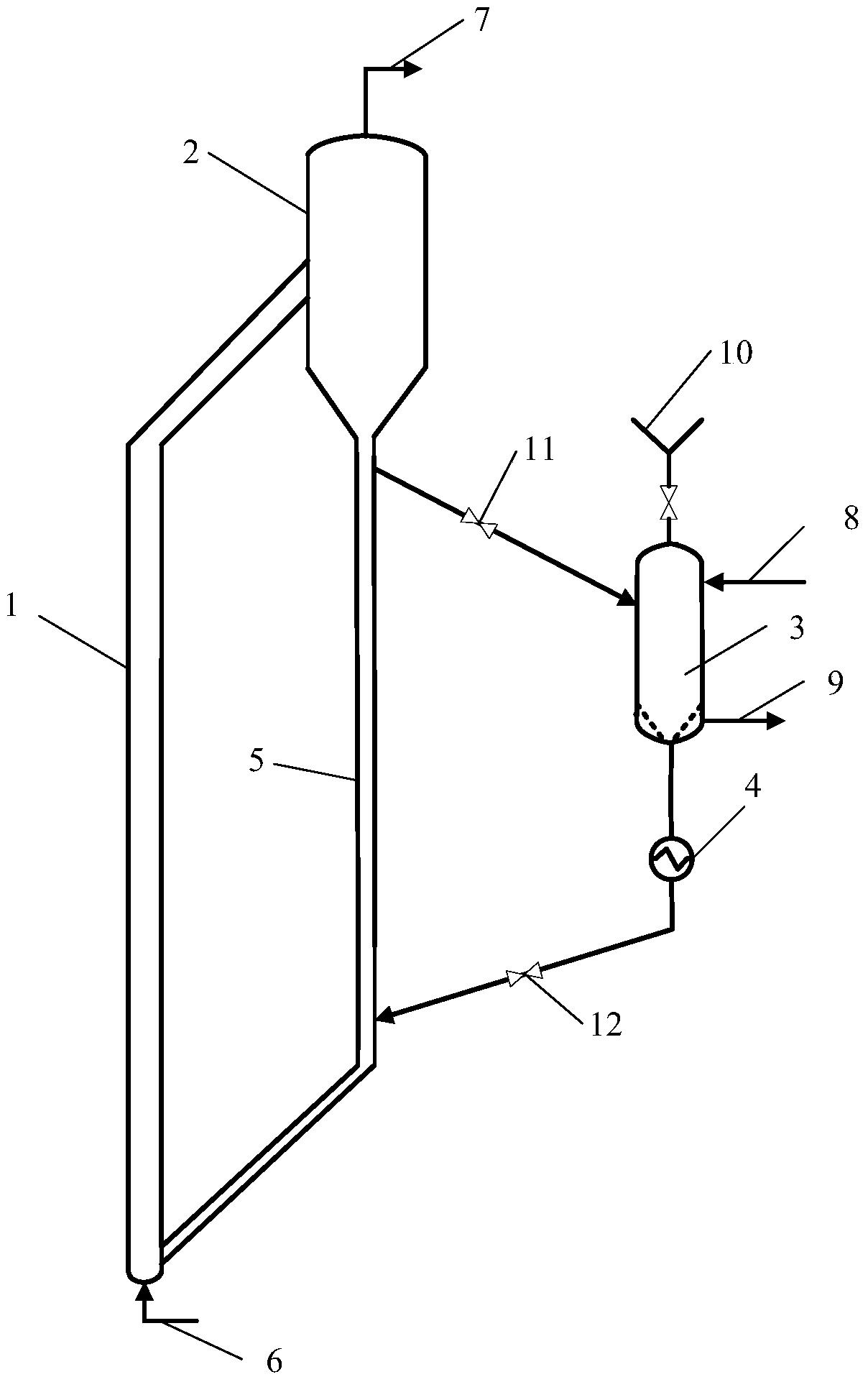
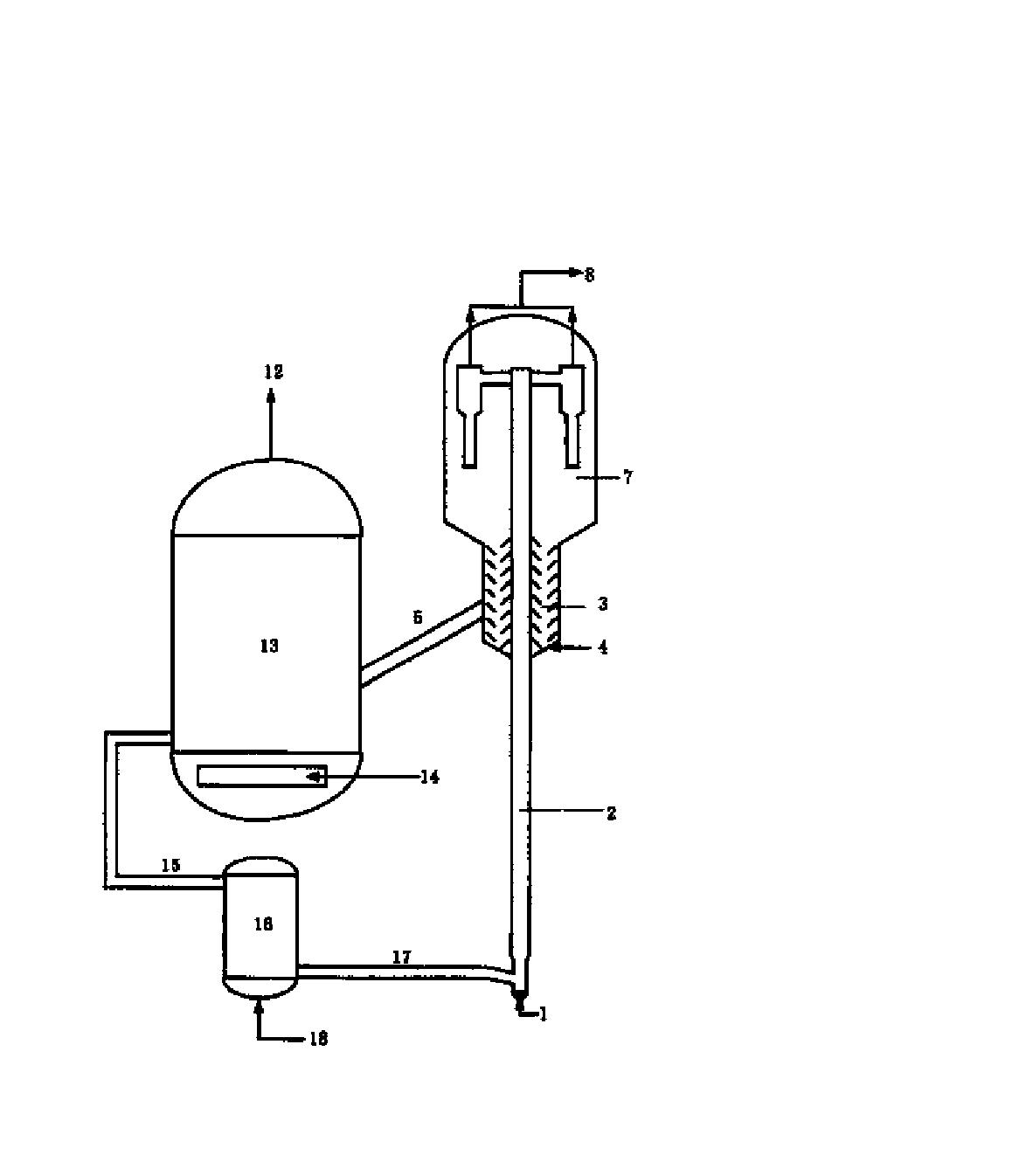
本发明提供的含酸油催化脱酸方法,将含酸烃油原料引入反应器,与保护剂接触,脱除原料油中的残碳前驱物,然后与催化脱酸催化剂接触进行脱酸反应得到酸值降低的烃油;所述的保护剂床层温度为200~380℃,所述的催化剂床层温度为200~380℃,反应器中水和二氧化碳的合并分压不大于350kPa、重时空速为0.1~50hr-1。
本发明提供的方法中,所述的保护剂选自氧化铝、累托石、蒙脱土、天然硅藻土和硅铝胶中的一种或几种。优选保护剂的平均孔径为80~ 比表面积为120~400m2/g,孔容为0.40~1.0cm3/g。
本发明提供的方法中,所述的催化脱酸催化剂含有碱土金属氧化物,含或不含过渡金属氧化物,含或不含载体,当所述的催化脱酸催化剂含有载体时,可以制备成负载型催化剂,也可以经机械混合制备成复合型催化剂。以催化剂的总重量计,所述的脱酸催化剂中含有0.1~100wt%的碱土金属氧化物、0~50wt%的过渡金属氧化物和0~99.9wt%的载体。
所述的催化脱酸催化剂一个优选的方案是采用金属氧化物组分负载在载体上的负载型催化剂,催化剂由碱土金属氧化物和载体组成,含或不含过渡金属氧化物,以催化剂的总重量计,所述的催化脱酸催化剂含有0.1~20wt%的碱土金属氧化物、0~8wt%的过渡金属氧化物和80~99.9wt%的载体。
所述的催化脱酸催化剂另一个优选的方案是采用混合型催化剂,以催化剂的总重量计,所述的催化脱酸催化剂中含有60~100wt%的碱土金属氧化物、0~40wt%的过渡金属氧化物。
所述碱土金属氧化物优选为MgO和/或CaO,更优选纳米氧化镁;所述过渡金属为Ti、Zr、V、Cr、Mn、Fe、Co、Ni、Cu、Ag、Zn、稀土金属中的一种或几种,所述的载体是氧化铝、氧化硅、氧化锆。所述催化脱酸催化剂的物化性质优选为比表面积为50~200m2/g,平均孔径为50~
本发明提供的方法中,所述的催化剂床层的温度优选为300~350℃,所述的水和二氧化碳的合并分压优选不超过200kPa,更优选不超过100kPa。
本发明提供的方法中,所述的含酸烃油原料为酸值不小于0.5mgKOH/g的原油和/或馏分油,更优选酸值不小于1.5mgKOH/g的原油和/或馏分油。
本发明提供的方法的有益效果为:
由于含酸原油和馏分油中含有大量易于生焦的残炭前驱物,此类物质分子大,结构复杂,饱和度低,芳香性高,易于沉积在脱酸催化剂的孔道内堵塞孔道,使催化剂失活。本发明提供的含酸油脱酸方法,在脱酸催化剂前设置大孔径的保护剂,保护剂可以容纳较多的残炭前驱物,减少了这部分物质在脱酸催化剂上的吸附,使得脱酸催化剂具有较高的脱酸活性和稳定性。
附图说明具体实施方式具体实施方式
下面将通过实施例对本发明的效果作进一步说明,但不因此而限制本发明。
对比例1
对比例1说明现有技术中单独采用脱酸催化剂的脱酸效果。
(1)制备现有技术中的脱酸催化剂纳米氧化镁:
按照文献“新颖氧化镁纳米带共沉淀法合成与表征”(无机化学学报,2005,21(6))中的方法,取56g(NH4)2CO3溶于1200mL去离子水中,加热至70℃,量取80mL氨水,加入(NH4)2CO3溶液中形成混合沉淀剂;取64克Mg(NO3)2·6H2O溶于1200mL去离子水中,在搅拌情况下,将升温至70℃的混合沉淀剂快速倾倒入硝酸镁溶液中;40分钟后停止搅拌,在室温下陈化约30min,过滤、洗涤,滤饼80℃常压干燥,然后于500~700℃煅烧,得到10g纳米氧化镁,经压片成型,破碎成20~40目,记为C-1,组成和孔结构数据见表1。
(2)含酸原油脱酸实验
使用连续进料固定床微型反应装置评价含酸原油脱酸效果,反应器中装填10g催化剂C-1,通入酸值为7.5mgKOH/g的馏分油1,原料油性质见表2,反应温度为350℃,重时空速为1.0hr-1,用氮气吹扫维持水和CO2的合并分压为100kPa。进油1.0小时后取油样分析酸值,并计算脱酸率,以此作为催化剂的初始活性;取反应19.0~20.0小时的油样分析其酸值,并计算脱酸率,作为催化剂的稳定活性。C-1催化剂的初始活性为脱酸率83.0%,脱酸结果见表3。
注:脱酸率=(原料的TAN-反应后收集油样的TAN)×100/原料的TAN。
实施例1
实施例1说明本发明提供的方法的脱酸效果。
(1)制备保护剂
将拟薄水铝石与粘结剂按2∶1混合后挤条成型,经干燥、550℃焙烧后,破碎成20~40目,记为P-1,组成和孔结构数据见表1。
(2)装填保护剂的含酸原油脱酸实验
采用对比例1中的评价装置,反应器中上层装填10g保护剂P-1,下层装填10g脱酸催化剂C-1,原料为酸值为7.5mgKOH/g的馏分油1,原料油性质见表2,保护剂床层温度为250℃,重时空速为1.0hr-1,脱酸催化剂床层温度为350℃,重时空速为1.0hr-1,用氮气吹扫维持水和CO2的合并分压为100kPa。按照对比例1中的方法计算催化剂初始活性和稳定活性,初始活性为脱酸率98.2%,脱酸结果见表3。
实施例2
实施例2说明本发明提供的方法的脱酸效果。
(1)制备保护剂
取100ml水玻璃(长岭炼油化工厂催化剂厂产品,比重1.26,SiO2含量250g/L,模数3.25)放入一烧杯中,加入54mL高偏铝酸钠溶液(长岭炼油化工厂催化剂厂产品,比重1.33,Al2O3含量41g/L,Na2O含量297g/L),混合均匀后,于室温老化24小时,然后取其220g,215ml硫酸铝溶液(长岭炼油化工厂催化剂厂产品,比重1.28,Al2O3含量102g/L,Na2O含量155.1g/L)及280ml去离子水,加到6401水玻璃中,加完后继续强烈搅拌1小时,然后将凝胶在60℃老化20小时。制备硅铝胶后,破碎成20~40目,记为P-2,组成和孔结构数据见表1。
(2)装填保护剂P-2的含酸原油催化剂催化脱酸实验
采用实施例1中的反应装置和评价方法进行含酸原油催化脱酸实验,反应器中上层装填10g保护剂P-2,下层装填脱酸催化剂10g C-1,原料为酸值为7.5mgKOH/g的馏分油1,原料油性质见表2,保护剂床层温度为250℃,重时空速为40.0hr-1,脱酸催化剂床层温度为350℃,重时空速为40hr-1,用氮气吹扫维持水和CO2的合并分压为180kPa。按照对比例1中的方法计算催化剂初始活性和稳定活性,脱酸结果见表3。初始活性为脱酸率97.6%。
对比例2
对比例2说明现有技术中单独采用脱酸催化剂氧化钙/氧化铬的脱酸效果。
(1)制备脱酸催化剂
取9.0g氧化钙和1.0g氧化铬,机械混合均匀,压片成型,破碎成20~40目,记为C-2,组成和孔结构数据见表1。
(2)含酸原油催化脱酸实验
采用对比例1中的连续进料的固定床微型反应装置进行含酸原油催化脱酸实验,反应器中装填10g脱酸催化剂C-2,所用原料是酸值为4.6mgKOH/g的馏分油2,原料油性质见表2,反应温度为300℃,重时空速为5.0hr-1,用氮气吹扫维持水和CO2的合并分压为150kPa。按照对比例1中的方法计算催化剂初始活性和稳定活性,脱酸结果见表3。
实施例3
实施例3说明本发明提供的方法的脱酸效果。
所用评价装置和方法和实施例1相同,反应器中上层装填10g保护剂P-2,下层装填脱酸催化剂10g C-2,原料为酸值为4.6mgKOH/g的馏分油2,原料油性质见表2,保护剂床层温度为300℃,重时空速为10.0hr-1,脱酸催化剂床层温度为300℃,重时空速为10.0hr-1,用氮气吹扫维持水和CO2的合并分压为150kPa。按照对比例1中的方法计算催化剂初始活性和稳定活性,组合催化剂的初始活性为脱酸率95.7%,脱酸结果见表3。
对比例3
对比例3说明现有技术中单独采用脱酸催化剂氧化镁/氧化铝的脱酸效果。
(1)制备脱酸催化剂
将拟薄水铝石和粘结剂制备的P-1用饱和浸渍法在其上负载16%(以载体重量计)氧化镁,记为MA-1,组成和孔结构数据见表1。
(2)含酸馏分油催化脱酸实验
反应装置和评价方法同对比例1,反应器中装填10g催化剂MA-1,原原料为酸值为10.2mgKOH/g的馏分油3,原料油性质见表2,反应温度为250℃,重时空速为40.0hr-1,用氮气吹扫维持水和CO2的合并分压为200kPa。按照对比例1中的方法计算催化剂初始活性和稳定活性,MA-1催化剂的初始活性为脱酸率81.7%,脱酸结果见表4。
实施例4
实施例4说明本发明提供的方法的脱酸效果。
采用对比例1中的反应装置和评价方法,反应器中上层装填10g保护剂P-1,下层装填脱酸10g催化剂MA-1,原料是酸值为10.2mgKOH/g的馏分油3,原料油性质见表2,保护剂床层温度为350℃,重时空速为20.0hr-1,脱酸催化剂床层温度为250℃,重时空速为20.0hr-1,用氮气吹扫维持水和CO2的合并分压为200kPa。按照对比例1中的方法计算催化剂初始活性和稳定活性,组合催化剂的初始活性为脱酸率99.1%,脱酸结果见表4。
对比例4
对比例3说明现有技术中单独采用脱酸催化剂氧化钙/氧化硅的脱酸效果。
(1)制备脱酸催化剂
以氧化硅为载体,用饱和浸渍法在其上负载8.5%(以载体重量计)的氧化钙和2.1%(以载体重量计)的氧化银,记为MA-2,组成和孔结构数据见表1。
(2)含酸馏分油催化脱酸实验
采用对比例1中的反应装置和评价方法,反应器中装填10g催化剂MA-2,所用原料是酸值为5.3mgKOH/g的脱水脱盐原油A,原料油性质见表2,反应温度为350℃,重时空速为20.0hr-1,用氮气吹扫维持水和CO2的合并分压为300kPa。按照对比例1中的方法计算催化剂初始活性和稳定活性,MA-2催化剂的初始活性为脱酸率69.0%,脱酸结果见表4。
实施例5
实施例5说明本发明提供的含酸原油脱酸方法的脱酸效果。
采用对比例1中的反应装置和评价方法,反应器中上层装填10g保护剂P-2,下层装填脱酸10g催化剂MA-2,所用原料是酸值为5.3mgKOH/g的脱盐脱水原油A,原料油性质见表2,保护剂床层温度为300℃,重时空速为30.0hr-1,脱酸催化剂床层温度为350℃,重时空速为30hr-1,用氮气吹扫维持水和CO2的合并分压为300kPa,按照对比例1中的方法计算催化剂初始活性和稳定活性,组合催化剂的初始活性为脱酸率96.2%,脱酸结果见表4。
表1催化剂和保护剂组成和物性
表2原料油性质
表3
表4
由表3可见,实施例1、2在反应器脱酸催化剂床层上分别装填了保护剂床层P-1、P-2,和对比例1只装填脱酸催化剂床层的方法相比,采用同一种脱酸催化剂,处理同样原料的初活脱酸率提高了14.6~15.2个百分点,稳定活性脱酸率提高了57.3~61.3个百分点;实施例3在脱酸催化剂床层上装填了保护剂P-2,和对比例2相比,初活脱酸率提高了20.5个百分点,稳定活性脱酸率提高了63.7个百分点;实施例4在脱酸催化剂MA-1上层装填了保护剂P-1,和对比例3相比,初活脱酸率提高了17.4个百分点,稳定活性脱酸率提高了35.8个百分点;实施例5在催化剂脱酸催化剂MA-2上层装填了保护剂P-2,和对比例4相比,初活脱酸率提高了27.2个百分点,稳定活性脱酸率提高了33.7个百分点;可见,采用本发明提供的方法处理含酸烃油原料,和现有技术中直接采用脱酸催化剂处理含酸烃油原料的方法相比,脱酸率都有了显著的提高,特别是随反应时间进行,脱酸率下降缓慢,从而延长了脱酸催化剂的使用寿命。
一种含酸油催化脱酸方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0