

IPC分类号 : C07J9/00,C07J73/00,C07J17/00,C07J41/00,C07J71/00,A61K31/575,A61K31/58,A61K31/585,A61P27/12







专利摘要
本发明公开了一种化合物及其在治疗白内障中的应用。所述化合物的结构式如式Ⅰ所示。式Ⅰ所示化合物、其前药或其药学上可接受的盐可用于阻止、缓解或者逆转晶状体蛋白在细胞内的聚集;在晶状体细胞中,90%以上的蛋白组分是晶状体蛋白(crystallin,CRY),包括α‑、β‑和γ‑CRY三个家族,而晶状体蛋白发生突变后,会引发细胞内的蛋白聚集,导致白内障疾病,本发明将选取α‑CRY家族突变体αA‑Y118D、αB‑R120G、β‑CRY家族突变体βB2‑V187E、γ‑CRY家族突变体γC‑G129C和γD‑W43R为白内障疾病的研究模型检测了本发明化合物的效果。本发明提供的具有新颖结构的小分子,与现有的小分子(如C29,Science,350,674)相比,在抑制细胞内晶状体蛋白突变导致的蛋白聚集具有更好的活性,且提高药物的可被机体吸收性,并且对正常晶状体细胞没有毒副作用。
权利要求
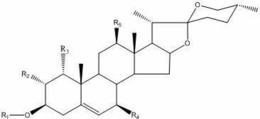
1.式Ⅰ所示化合物、其前药或其药学上可接受的盐,
式Ⅰ中,环A为六元环或七元环,当环A为六元环时,X不存在,当环A为七元环时,X表示O或NH;
3号碳原子与R1之间为单键或双键,当为单键时,R1选自如下基团中任一种:
当为双键时,R1选自如下基团中任一种:
7号碳原子、8号碳原子、9号碳原子和11号碳原子相邻碳原子之间为单键或双键;
当8号碳原子与9号碳原子之间为单键时,8号碳原子与9号碳原子之间存在环氧结构或7号碳原子与8号碳原子之间为双键或9号原子与11号碳原子之间为双键;
当7号碳原子与8号碳原子之间为单键时,R2为H或2个R2形成羰基;
当7号碳原子与8号碳原子之间为双键时,7号碳原子上连接一个R2,R2为H;
当9号碳原子与11号碳原子之间为单键时,R3为H或2个R3形成羰基;
当9号碳原子与11号碳原子之间为双键时,11号碳原子上连接一个R3,R3为H;
R4选自如下基团中任一种:
其中,R5和R6均为碳原子数为1-4的烷基;
X”和X”’均选自F、Cl、Br和I;
n为1或2。
2.根据权利要求1所述的化合物、其前药或其药学上可接受的盐,其特征在于:所述化合物的结构式如式Ⅱ所示,
式Ⅱ中,R1、R2、R3、R4和n的定义同式Ⅰ中。
3.根据权利要求2所述的化合物、其前药或其药学上可接受的盐,其特征在于:所述化合物的结构式如式Ⅲ所示,
式Ⅲ中,R’4选自如下基团中任一种:
4.根据权利要求2所述的化合物、其前药或其药学上可接受的盐,其特征在于:所述化合物的结构式如式Ⅳ所示,
式Ⅳ中,R’1与碳原子之间为单键或双键;
当为单键时,R’1选自如下基团中任一种:
当为双键时,R’1选自如下基团中任一种:
R”4选自如下基团中任一种:
5.根据权利要求2所述的化合物、其前药或其药学上可接受的盐,其特征在于:所述化合物的结构式如式Ⅴ所示,
式Ⅴ中,R”1与碳原子之间为单键或双键;
当为单键时,R”1为-OH;
当为双键时,R”1为
6.根据权利要求2所述的化合物、其前药或其药学上可接受的盐,其特征在于:所述化合物的结构式如式Ⅵ所示,
7.根据权利要求1所述的化合物、其前药或其药学上可接受的盐,其特征在于:所述化合物的结构式如式Ⅶ所示,
式Ⅶ中,X表示O或NH。
8.权利要求1-7中任一项所述化合物、其前药或其药学上可接受的盐在制备治疗白内障的药物中的应用。
9.权利要求1-7中任一项所述化合物、其前药或其药学上可接受的盐在制备阻止、缓解或逆转晶状体蛋白在细胞内聚集的产品中的应用。
10.一种治疗白内障的药物,其活性成分为权利要求1-7中任一项所述化合物、其前药或其药学上可接受的盐。
说明书
技术领域
本发明涉及一种化合物及其在治疗白内障中的应用,属于生物医药领域。
背景技术
晶状体是眼球屈光系统的重要组成部分,也是唯一具有调节能力的屈光间质,晶状体由晶体囊、晶体上皮、晶体纤维和悬韧带组成。如果晶状体由于各种原因造成其部分或全部混浊,则发生白内障。白内障会导致单眼或者双眼的视力降低。通常白内障发展缓慢,症状主要包括视力模糊、晕光、夜视能力降低,严重会致盲。视力的降低会严重的影响人们的日常生活,比如开车、阅读,视力的降低也会导致心理疾病的产生。白内障多发于40岁以上的人群,随着且随年龄增长而增多,与多因素相关,如与老年人代谢缓慢,发生退行性病变有关,也有人认为与日光长期照射,内分泌紊乱,代谢障碍等因素有关。外伤、药物、放射性物质、并发症等也会引起后天性白内障,另外,有一些先天性的白内障患者,多在出生前后即已存在,有内生性与外生性两类,内生性者与胎儿发育障碍有关,外生性者是母体或胎儿的全身病变对晶状体造成损害所致。
国际公认的快速有效治疗白内障方法是手术治疗,通过手术将患者浑浊的晶状体取出,然后植入人工晶体。但总体而言手术治疗费用较高,对患者而言是很大的经济负担,随着人类平均寿命的延长、人口老龄化的出现,这一难题更为突出,因此寻找有效、安全、廉价的药物治疗白内障具有重要的现实意义。
晶状体内90%的蛋白是由晶状体蛋白组成。其中α-、β-和γ-晶状体蛋白是晶状体内最主要的可溶性蛋白。其中,α-晶状体蛋白是由两个亚基组成的二聚体,属于小热休克蛋白家族,它可以在不依赖ATP的情况下有效的结合损伤的或者未能正确折叠的蛋白质从而阻止这些蛋白的聚集。从白内障患者的晶状体中可以分离得到的很多不同种类的蛋白,其中有很多是以高分子量的蛋白聚集体形式存在。这些蛋白聚集体导致了整个白内障晶状体的浑浊遮光性。因此需要提供一种可以结合晶状体蛋白的药物以治疗白内障。
发明内容
本发明的目的是提供一种新具有新颖结构的小分子化合物,该小分子化合物在抑制晶状体细胞内蛋白聚集方面具有更好的活性,并且对正常晶状体细胞没有毒副作用。
本发明首先提供式Ⅰ所示化合物、其前药或其药学上可接受的盐,
式Ⅰ中,环A为六元环或七元环,当环A为六元环时,X不存在,当环A为七元环时,X表示O或NH;
3号碳原子与R1之间为单键或双键,当为单键时,R1选自如下基团中任一种:
当为双键时,R1选自如下基团中任一种:
7号碳原子、8号碳原子、9号碳原子和11号碳原子相邻碳原子之间为单键或双键;
当8号碳原子与9号碳原子之间为单键时,8号碳原子与9号碳原子之间存在环氧结构或7号碳原子与8号碳原子之间为双键或9号原子与11号碳原子之间为双键;
当7号碳原子与8号碳原子之间为单键时,R2为H或2个R2形成羰基;
当7号碳原子与8号碳原子之间为双键时,7号碳原子上连接一个R2,R2为H;
当9号碳原子与11号碳原子之间为单键时,R3为H或2个R3形成羰基;
当9号碳原子与11号碳原子之间为双键时,11号碳原子上连接一个R3,R3为H;
R4选自如下基团中任一种:
其中,R5和R6均为碳原子数为1-4的烷基;
X”和X”’均选自F、Cl、Br和I;
n为1或2。
所述化合物的结构式进一步如式Ⅱ所示,
式Ⅱ中,R1、R2、R3、R4和n的定义同式Ⅰ中。
所述化合物的结构式具体如式Ⅲ所示,
式Ⅲ中,R’4选自如下基团中任一种:
式Ⅲ所示化合物具体如式Ⅲ-1、式Ⅲ-2、式Ⅲ-3、式Ⅲ-4、式Ⅲ-5、式Ⅲ-6、式Ⅲ-7、式Ⅲ-8或式Ⅲ-9所示:
所述化合物的结构式具体如式Ⅳ所示,
式Ⅳ中,R’1与碳原子之间为单键或双键;
当为单键时,R’1选自如下基团中任一种:
当为双键时,R’1选自如下基团中任一种:
R”4选自如下基团中任一种:
式Ⅳ所示化合物具体如式Ⅳ-1、式Ⅳ-2、式Ⅳ-3、式Ⅳ-4、式Ⅳ-5、式Ⅳ-6、式Ⅳ-7、式Ⅳ-8、式Ⅳ-9、式Ⅳ-10、式Ⅳ-11、式Ⅳ-12或式Ⅳ-13所示:
所述化合物的结构式具体如式Ⅴ所示,
式Ⅴ中,R”1与碳原子之间为单键或双键;
当为单键时,R”1为-OH;
当为双键时,R”1为
式Ⅴ所示化合物具体如式Ⅴ-1或式Ⅴ-2所示:
所述化合物的结构式具体如式Ⅵ所示,
所述化合物的结构式进一步如式Ⅶ所示,
式Ⅶ中,X表示O或NH。
式Ⅶ所示化合物具体如式Ⅶ-1或式Ⅶ-2所示:
本发明化合物可根据现有的常规方法进行制备,如采用氧化反应、还原反应和/或缩合反应等常规反应进行。
本发明所提供的化合物、其前药或其药学上可接受的盐可用于治疗白内障。
本发明提供的化合物、其前药或其药学上可接受的盐可用于阻止、缓解或者逆转晶状体蛋白在细胞内的聚集;
在晶状体细胞中,90%以上的蛋白组分是晶状体蛋白(crystallin,CRY),包括α-、β-和γ-CRY三个家族,而晶状体蛋白发生突变后,会引发细胞内的蛋白聚集,导致白内障疾病,本发明将选取α-CRY家族突变体αA-Y118D、αB-R120G、β-CRY家族突变体βB2-V187E、γ-CRY家族突变体γC-G129C和γD-W43R为白内障疾病的研究模型来检测本发明化合物的效果。
活性成分为本发明提供的化合物、其前药或其药学上可接受的盐的治疗白内障的药物也属于本发明的保护范围。
本发明提供的具有新颖结构的小分子,与现有的小分子(如C29,Science,350,674)相比,在抑制细胞内晶状体蛋白突变导致的蛋白聚集具有更好的活性,且提高药物的可被机体吸收性,并且对正常晶状体细胞没有毒副作用。
附图说明
图1为晶状体蛋白突变体αB R120G在细胞内发生错误折叠形成聚集小体的示意图。
图2为本发明化合物对多种晶状体蛋白突变体形成聚集体的效应。
图3为本发明式Ⅲ-6所示化合物对晶状体蛋白突变体αB R120G聚集的半效应浓度。
图4为本发明式Ⅲ-6所示化合物细胞毒性的检测结果。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1、式Ⅲ-1所示化合物的制备
羊毛甾醇(50%,购于TCI,100mg)溶于二氯甲烷(30mL),在冰浴下,m-CPBA(间氯过氧苯甲酸)(85%,28mg)和NaHCO3(14mg)间隔3h分两批加入到上述溶液中,室温搅拌过夜。反应液用饱和碳酸氢钠溶液洗涤,无水碳酸钠干燥,然后再旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=10:1,体积比),得式Ⅲ-1所示化合物(45mg)和式Ⅲ-1’所示化合物(40mg)。
式Ⅲ-1’所示化合物:1H-NMR(400MHz,CDCl3)δ(ppm)3.23-3.21(m,1H),0.68(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)134.5,79.1,50.7,50.6,49.9,44.6,39.7,39.0,37.2,36.6,35.7,31.1,31.0,29.9,28.4,28.1,28.0,26.7,24.4,24.3,23.0,22.7,21.2,19.3,18.9,18.4,15.9,15.6.
式Ⅲ-1所示化合物:1H-NMR(400MHz,CDCl3)δ(ppm)3.21(d,J=8.0Hz,1H),2.69-2.67(m,1H),0.68(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)134.6,134.4,79.0,77.5,77.2,76.8,65.0,64.9,58.5,58.2,50.51,50.48,50.4,49.9,44.6,39.0,37.1,36.4,36.3,35.7,32.9,32.7,31.10.31.08,30.9,28.4,28.3,28.1,28.0,26.6,26.0,25.7,25.06,25.05,24.4,21.1,19.3,18.9,18.79,18.76,18.7.
经表征,所制备的化合物结构正确。
实施例2、式Ⅲ-2所示化合物的制备
高碘酸钠(135mg)加入到35mL的乙醚中,搅拌的条件下,式Ⅲ-1所示化合物(283mg)加入到上述反应液中,室温下搅拌15min,水加入到反应液中,有机层采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=15:1,体积比),得式Ⅲ-2所示化合物(120mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)9.76(s,1H),3.24-3.21(m,1H),2.46-2.35(m,2H),0.6(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)203.4,134.6,134.4,79.1,77.5,77.2,76.9,53.6,50.5,50.4,50.0,44.7,41.3,39.0,37.2,36.2,35.7,31.1,30.9,28.4,28.3,28.1,28.0,26.6,24.4,21.1,19.3,18.5,18.4,15.9,15.6.
经表征,所制备的化合物结构正确。
实施例3、式Ⅲ-3所示化合物的制备
羊毛甾醇(50%,购于TCI,220mg)溶于THF/H2O(20mL/5mL),NBS(54mg)加入到反应液中,反应液在室温下搅拌2h。反应液加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=5:1,体积比),得式Ⅲ-3所示化合物(110mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)4.13-3.94(m,1H),3.25-3.21(m,1H),0.69(s,3H).
经表征,所制备的化合物结构正确。
实施例4、式Ⅲ-4所示化合物的制备
将40mg式Ⅲ-2所示化合物,25mg NaOAc,14mg NH2OH·HCl溶于5ml 1,4-二氧六环-水(v/v,2:1)中,置于室温下搅拌12h。TLC检测其反应完全,将反应产物用饱和食盐水洗涤,乙酸乙酯萃取。分离有机相,用无水硫酸钠干燥后,置于旋转蒸发仪上减压蒸干。采用硅胶柱(v/v,乙酸乙酯:石油醚=1:5)分离纯化,得到式Ⅲ-4所示化合物17mg。
1H-NMR(400MHz,CDCl3)δ(ppm)3.24(dd,J=11.6Hz,J=4.4Hz),0.88(s,3H),0.81(s,3H),0.69(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)153.1,134.6,134.5,79.2,50.6,50.5,50.4,50.0,44.7,39.1,37.2,36.5,36.3,35.8,33.1,32.6,31.1,31.0,29.9,28.4,28.3,28.1,28.0,26.7,24.4,21.2,19.3,18.54,18.48,18.4,15.9,15.6.
经表征,所制备的化合物结构正确。
实施例5、式Ⅲ-5所示化合物的制备
将40mg式Ⅲ-2所示化合物,3ml二乙胺溶于二氯乙烷中搅拌1h后,于0℃下缓慢加入42mg NaBH(OAc)3。恢复至室温并继续搅拌12h。加入少许酸猝灭反应后,用饱和碳酸氢钠洗涤,乙酸乙酯萃取。分离有机相,用无水硫酸钠干燥后,置于旋转蒸发仪上减压蒸干。采用硅胶柱(v/v,二氯甲烷:甲醇=20:1)分离纯化,得到式Ⅲ-5所示化合物40mg。
1H-NMR(400MHz,CDCl3)δ(ppm)3.21(dd,J=11.2Hz,J=3.6Hz,1H),2.27-2.55(m,4H),0.79(s,3H),0.67(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)134.54.134.48,79.0,53.5,50.53,50.48,49.9,46.9,44.6,39.0,37.1,36.5,35.7,34.2,31.1,31.0,28.3,28.1,28.0,26.6,24.4,23.4,21.1,19.3,18.9,18.4,15.9,15.6,11.5.
经表征,所制备的化合物结构正确。
实施例6、式Ⅲ-6所示化合物的制备
式Ⅲ-3所示化合物(80mg)溶于无水THF(10mL)中,LiAlH4(38mg)加入到反应液中,反应液加热回流2h。冷却到室温,加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=5:1,体积比),得式Ⅲ-6所示化合物(40mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)3.25-3.22(m,1H),2.03-1.89(m,5H),0.69(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)134.58,134.56,79.1,71.3,50.7,50.6,50.0,44.7,44.6,39.0,37.2,36.9,36.6,35.8,31.2,31.0,29.5,29.4,28.4,28.1,28.0,26.7,24.4,21.3,21.2,19.3,18.8,18.4,15.9,15.6.
经表征,所制备的化合物结构正确。
实施例7、式Ⅲ-7所示化合物的制备
将50mg式Ⅲ-2所示化合物溶于无水5ml THF中。加入4ml 4M异丙基溴化镁的四氢呋喃溶液。TLC检测反应完成后,加水终止反应,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=5:1,体积比),得式Ⅲ-7所示化合物17mg。
1H-NMR(400MHz,CDCl3)δ(ppm)3.33-3.32(m,1H),3.23(dd,J=11.6Hz,J=4.8Hz,1H),0.81(s,3H),0.69(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)134.54,134.51,79.1,50.6,50.5,44.6,39.0,37.2,35.7,31.1,31.0,28.1,28.0,27.0,26.6,24.4,22.8,21.1,19.3,19.2,19.0,18.4,16.8,15.9,15.6,14.3.
经表征,所制备的化合物结构正确。
实施例8、式Ⅲ-8所示化合物的制备
式Ⅲ-6所示化合物(50mg)于二氯甲烷(30mL),在冰浴下,m-CPBA(85%,28mg)和NaHCO3(14mg)间隔3h分两批加入到上述溶液中,室温搅拌过夜。反应液用饱和碳酸氢钠溶液洗涤,无水碳酸钠干燥,然后再旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=5:1,体积比),得式Ⅴ-1所示化合物(45mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)3.21-3.19(m,1H),0.76-0.75(m,6H)13C-NMR(100MHz,CDCl3)δ(ppm)78.6,71.2,70.8,68.3,49.0,48.5,44.5,43.7,41.9,38.6,38.0,36.7,36.4,33.0,29.42,29.36,28.6,28.4,27.3,27.0,23.6,21.6,21.2,20.1,19.1,17.1,16.6,16.4,15.2.
将30mg式Ⅴ-1所示化合物溶于3ml THF中,加入100μl 40%的氢氟酸溶液,室温下搅拌4天。TLC检测反应完成后用饱和碳酸氢钠溶液洗,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱(乙酸乙酯:石油醚=1:3)分离得到27mg式Ⅲ-8所示化合物。
1H-NMR(400MHz,CDCl3)δ(ppm)5.47(s,br,1H),5.32(s,br,1H),3.24(dd,J=11.2Hz,J=4.4Hz,1H),0.56(s,3H)13C-NMR(100MHz,CDCl3)δ(ppm)146.0,142.8,120.3,116.5,79.1,71.2,51.2,50.5,49.2,44.5,43.9,38.8,38.0,37.5,36.8,36.4,35.8,31.6,29.5,29.4,28.3,28.1,27.9,25.7,23.1,22.9,21.3,18.6,15.9,15.8.
经表征,所制备的化合物结构正确。
实施例9、式Ⅲ-9所示化合物的制备
式Ⅲ-6所示化合物(44mg)溶于无水二氯甲烷中,冰浴条件下下,20μl DAST加入到反应体系中,反应1h。加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=10:1),得式Ⅲ-9所示化合物(10mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)3.23(dd,J=11.6Hz,J=4.4Hz,1H),0.69(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)150.5,140.8,122.1,116.4,96.9,95.3,59.1,50.4,50.35,45.5,44.8,42.1,41.9,37.2,36.7,36.6,36.3,30.5,29.8,29.6,28.2,27.0,26.9,26.8,26.6,24.5,24.0,23.3,23.2,22.9,20.9,18.9,16.0,15.9.
经表征,所制备的化合物结构正确。
实施例10、式Ⅳ-1所示化合物的制备
式Ⅲ-1所示化合物(80mg),醋酸酐(300uL),DMAP(2mg),吡啶(1ml)和二氯甲烷(10ml)加入到反应瓶中。室温搅拌6h。加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯20:1),得式Ⅳ-1所示化合物(80mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)4.51-4.47(m,1H),2.68(t,J=6.4Hz,1H),0.69(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)171.2,134.5,134.4,81.0,77.5,77.2,76.8,65.1,64.9,58.5,58.3,50.6,50.5,50.4,49.9,44.6,37.9,37.0,36.5,36.3,35.4,32.9,32.7,31.07,31.05,30.9,28.4,28.3,28.0,26.5,26.1,25.7,25.09,25.07,24.4,24.3,21.5,21.1,19.3,18.9,18.8,18.7,18.2,16.7.
经表征,所制备的化合物结构正确。
实施例11、式Ⅳ-2所示化合物的制备
羊毛甾醇(80mg)、醋酸酐(300uL)、DMAP(2mg)、吡啶(1ml)和二氯甲烷
(10ml)加入到反应瓶中。室温搅拌6h。加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯20:1),得式Ⅳ-2所示化合物(80mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)5.12-5.08(m,1H),4.52-4.48(m,1H),0.68(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)171.0,134.7,134.5,131.0,125.4,81.1,77.5,77.2,76.9,50.7,50.6,50.0,44.7,38.0,37.1,36.5,36.4,35.5,31.2,31.0,28.4,28.1,26.6,25.8,25.1,24.4,24.37,21.4,21.2,19.3,18.8,18.3,17.8,16.7,15.9.
经表征,所制备的化合物结构正确。
实施例12、式Ⅳ-3所示化合物的制备
式IV-1(500mg)溶解到异丙醇中,水(5mL)和次磷酸(0.9mL,50%水溶液)加入到上述溶液中,反应液加热回流3小时,用水稀释,过滤得白色固体。产品用硅胶柱分离(二氯甲烷:乙酸乙酯2:1),得式Ⅳ-3所示化合物(450mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)4.51-4.47(m,1H),3.36-3.27(m,1H),0.68(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)171.2,134.6,134.4,81.1,79.8,78.9,73.4,73.3,50.7,50.6,49.9,44.6,37.9,37.0,36.9,36.4,35.4,33.7,33.2,31.1,30.9,28.8,28.5,28.4,28.3,28.0,26.7,26.5,24.4,24.3,23.4,23.3,21.5,21.1,19.3,19.0,18.7,18.2,16.7,15.9.
经表征,所制备的化合物结构正确。
实施例13、式Ⅳ-4所示化合物的制备
羊毛甾醇(100mg),PCC(100mg)和NaOAc(10mg)溶解到二氯甲烷中,室温搅拌1h。加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯20:1),得式Ⅳ-4所示化合物(90mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)5.11-5.08(m,1H),2.62-2.54(m,1H),2.43-2.37(m,1H),0.71(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)218.0,135.5,133.2,131.1,125.3,51.3,50.5,50.0,47.5,44.6,37.0,36.44,36.37,36.2,34.7,31.1,31.0,28.3,26.5,26.3,25.9,25.0,24.4,21.4,21.2,19.6,18.8,18.75,17.8,16.0.
经表征,所制备的化合物结构正确。
实施例14、式Ⅳ-5所示化合物的制备
式Ⅳ-4所示化合物(100mg),MeONH2·HCl(56mg),NaOAc(150mg)溶于乙醇(10ml),反应液60摄氏度加热搅拌3h。反应液冷却到室温,加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=10:1,体积比),得式Ⅳ-5所示化合物(95mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)9.11(s,br,1H),3.13(d,J=3.6Hz,1H),0.90(d,J=6Hz,3H),0.85(s,3H),0.70(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)167.2,134.9,133.9,71.3,51.5,50.6,50.0,44.6,44.5,40.5,37.3,36.8,36.6,35.8,31.1,31.0,29.5,29.3,28.3,27.0,26.5,24.4,23.1,21.3,21.2,19.1,18.9,18.8,17.7,16.0.
经表征,所制备的化合物结构正确。
实施例15、式Ⅳ-6所示化合物的制备
式Ⅳ-4所示化合物(100mg),HONH2·HCl(48mg),NaOAc(150mg)溶于乙醇(10ml),反应液60摄氏度加热搅拌3h。反应液冷却到室温,加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯10:1),得式1所示化合物。
1H-NMR(400MHz,CDCl3)δ(ppm)9.14(s,1H),5.10(s,1H),3.14-3.10(m,1H),0.70(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)167.2,135.0,134.0,131.0,125.4,51.5,50.5,50.0,44.6,40.5,37.3,36.5,36.4,35.8,31.1,31.0,28.3,27.1,26.5,25.9,25.1,24.4,23.2,21.2,19.1,18.9,18.8,17.8,17.7,16.0
式1所示化合物(50mg)溶于无水乙醚中,LiAlH4(50mg)加入到反应液中,室温反应3h。慢慢加入水来终止反应。用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(二氯甲烷:甲醇10:1),得式Ⅳ-6所示化合物(20mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)8.25(s,2H),5.10(m,1H),2.90(d,J=10.0Hz,1H),0.68(s,3H).
经表征,所制备的化合物结构正确。
实施例16、式Ⅳ-7所示化合物的制备
式Ⅲ-6所示化合物(100mg),PCC(100mg)和NaOAc(10mg)溶解到二氯甲烷中,室温搅拌1h。加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯20:1),得式Ⅳ-7所示化合物(90mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)2.56-2.40(m,2H),0.88(s,6H),0.70(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)217.9,135.47,133.28,71.17,51.4,50.6,50.0,47.5,44.6,44.5,37.0,36.9,36.6,36.2,34.7,31.1,31.0,29.5,29.4,28.3,26.5,26.3,24.4,21.4,21.2,19.6,18.8,16.0.
经表征,所制备的化合物结构正确。
实施例17、式Ⅳ-8所示化合物的制备
式Ⅳ-7所示化合物(100mg),HONH2·HCl(48mg),NaOAc(150mg)溶于乙醇(10ml),反应液60摄氏度加热搅拌3h。反应液冷却到室温,加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=10:1,体积比),得式Ⅳ-8所示化合物(95mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)9.11(s,br,1H),3.14-3.10(m,1H),0.91-0.89(m,6H),0.85(s,3H),0.70(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)167.2,134.9,133.9,71.3,51.5,50.6,50.0,44.6,44.5,40.5,37.3,36.8,36.6,35.8,31.1,31.0,29.5,29.3,28.3,27.0,26.5,24.4,23.2,21.3,21.2,19.1,18.9,18.8,17.7,15.9.
经表征,所制备的化合物结构正确。
实施例18、式Ⅳ-9所示化合物的制备
将40mg式Ⅲ-6所示化合物溶于4ml吡啶中,加入12mg丁二酸酐和11mg DMAP。置于80℃下搅拌3h。TLC检测反应完成后用10%的盐酸洗去吡啶,并用乙酸乙酯萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱(二氯甲烷:甲醇=20:1,体积比)分离得到31mg式Ⅳ-9所示化合物。
1H-NMR(400MHz,CDCl3)δ(ppm)4.52(dd,J=11.6Hz,J=4.8Hz,1H),2.69-2.63(m,4H),0.68(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)172.0,170.7,134.6,134.3,81.6,71.4,60.6,50.6,49.9,44.6,44.5,38.0,37.0,36.8,36.6,35.3,31.1,30.9,29.5,29.4,29.3,29.1,28.5,28.4,28.0,26.5,24.4,24.2,21.3.21.2,21.1,19.3,18.8,18.2,16.7.
经表征,所制备的化合物结构正确。
实施例19、式Ⅳ-10所示化合物的制备
式1所示化合物(50mg)溶于无水乙醚中,LiAlH4(50mg)加入到反应液中,室温反应3h。慢慢加入水来终止反应。用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(二氯甲烷:甲醇:三乙胺=10:1:0.1,体积比),得式Ⅳ-11所示化合物(20mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)2.68(s,1H),2.46-2.43(m,1H),0.69(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)134.6,134.2,70.1,68.4,59.7,51.1,50.9,50.5,49.7,44.3,43.9,38.3,37.04,36.97,36.9,36.7,36.4,35.8,35.6,31.0,30.9,30.5,27.9,27.7,27.0,26.3,25.4,25.2,23.3,20.69,20.65,18.4,18.13,18.07.
经表征,所制备的化合物结构正确。
实施例20、式Ⅳ-11所示化合物的制备
式Ⅳ-7所示化合物(100mg)、MeONH2·HCl(56mg)、NaOAc(150mg)溶于乙醇(10ml),反应液60摄氏度加热搅拌3h。反应液冷却到室温,加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯10:1),得式Ⅳ-12所示化合物(95mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)3.81(s,3H),3.00(d,J=14.4Hz,1H),0.70(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)166.2,134.9,134.0,77.5,77.2,76.8,71.3,61.2,51.6,50.7,50.0,44.62,44.56,40.2,37.2,36.9,36.6,35.8,31.1,31.0,29.5,29.4,28.4,27.2,26.5,24.4,23.4,21.3,21.2,19.1,18.8,18.3,16.0.
经表征,所制备的化合物结构正确。
实施例21、式Ⅳ-12和式Ⅳ-13所示化合物的制备
式Ⅲ-6所示化合物(90mg)溶于无水THF(10mL)中,NaH(5个当量)和碘甲烷(5个当量)加入到反应液中。室温搅拌12h。反应完毕后,逐滴加入水来终止反应。二氯甲烷加入到反应液中,有机相用无水硫酸钠干燥,然后旋转蒸发仪蒸干。所得产品采用硅胶柱分离(石油醚:乙酸乙酯=30:1)得式Ⅳ-13所示化合物(30mg)和式Ⅳ-14所示化合物(30mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)3.36(s,3H),3.69-3.65(m,1H),0.69(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)134.7,134.5,88.8,77.5,77.2,76.8,74.8,57.7,51.1,50.7,50.0,49.2,44.6,40.5,39.0,37.2,37.0,36.6,35.6,31.2,31.0,28.4,28.1,26.6,25.2,24.4,22.8,21.2,20.7,19.3,18.9,18.3,16.3,15.9.
1H-NMR(400MHz,CDCl3)δ(ppm)3.37(s,3H),3.17(s,3H),2.69-2.65(m,1H),0.68(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)134.7,134.5,88.8,71.3,57.7,51.1,50.7,50.0,44.7,44.6,39.0,37.2,36.9,36.6,35.7,31.2,31.0,29.5,29.4,28.4,28.1,26.6,24.4,22.8,21.3,21.2,19.3,18.8,18.3,16.3,15.9.
经表征,所制备的化合物结构正确。
实施例22、式Ⅴ-1所示化合物的制备
式Ⅲ-6所示化合物(50mg)溶于二氯甲烷(30mL),在冰浴下,m-CPBA(85%,28mg)和NaHCO3(14mg)间隔3h分两批加入到上述溶液中,室温搅拌过夜。反应液用饱和碳酸氢钠溶液洗涤,无水碳酸钠干燥,然后再旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=5:1,体积比),得式Ⅴ-1所示化合物(45mg)
1H-NMR(400MHz,CDCl3)δ(ppm)3.21-3.19(m,1H),0.76-0.75(m,6H)13C-NMR(100MHz,CDCl3)δ(ppm)78.6,71.2,70.8,68.3,49.0,48.5,44.5,43.7,41.9,38.6,38.0,36.7,36.4,33.0,29.42,29.36,28.6,28.4,27.3,27.0,23.6,21.6,21.2,20.1,19.1,17.1,16.6,16.4,15.2.
经表征,所制备的化合物结构正确。
实施例23、式Ⅴ-2所示化合物的制备
将37mg式Ⅴ-1所示化合物溶于3ml二氯甲烷,加入37mg PCC和4mg NaOAc,室温下搅拌2h。TLC检测反应完成后水洗,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离。将分离产物溶于乙醇,加入12mg盐酸羟胺和20mg醋酸钠。60℃搅拌2h。TLC检测反应完成后水洗,先用旋转蒸发仪蒸发除去大部分溶剂后,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱(乙酸乙酯:石油醚=5:1,体积比)分离得到21mg式Ⅴ-2所示化合物。
1H-NMR(400MHz,CDCl3)δ(ppm)3.14-3.10(m,1H),1.00(d,J=6Hz,3H),0.85(s,3H),0.70(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)166.4,71.3,70.7,68.3,49.0,48.5,44.5,43.7,43.1,40.0,38.1,36.7,36.4,32.9,32.1,29.44,29.38,28.6,27.6,26.9,23.9,22.8,21.7,21.2,20.1,19.1,17.4,17.2,17.0,16.4.
经表征,所制备的化合物结构正确。
实施例24、式Ⅵ所示化合物的制备
5H(30mg)、RuCl3·3H2O(1mg)和TBHP(indecane,5-6M)加入到2mL的环己烷中。室温反应6h。旋转蒸发仪旋干,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=5:1,体积比),得到的化合物溶于10%KOH的乙醇溶液中,回流3h,冷却至室温,加水稀释,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯=3:1,体积比,得式Ⅵ所示化合物C(10mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)3.29-3.25(m,1H),2.89(d,J=14.0Hz,1H),2.75(d,J=14.0Hz,1H),2.60(d,J=14.0Hz,1H),2.50-2.44(m,2H),0.80(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)202.6,202.4,151.9,150.8,77.8,71.2,51.8,50.3,49.3,49.1,47.5,44.4,39.9,39.0,36.6,36.3,34.2,32.3,29.5,29.4,28.0,27.8,27.5,26.0,21.1,18.7,17.7,17.0,15.6.
经表征,所制备的化合物结构正确。
实施例25、式Ⅶ-1所示化合物的制备
式Ⅳ-8所示化合物(50mg)溶于二氯甲烷/三氟乙酸(1mL/1ml)的混合溶剂中,室温反应2h。加水稀释,用二氯甲烷萃取。饱和碳酸氢钠洗涤。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱分离(石油醚:乙酸乙酯30:1),得式Ⅶ-1所示化合物5F(20mg)。
1H-NMR(400MHz,CDCl3)δ(ppm)8.72(s,1H),3.12(d,J=15.2Hz,1H),2.19-2.10(m,1H),0.72(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)167.3,135.0,134.0,89.5,51.5,50.6,50.0,44.6,40.9,40.5,37.3,36.43,36.38,35.8,31.1,31.0,28.3,27.1,26.5,25.8,25.7,24.4,23.2,21.2,20.6,19.1,18.9,18.7,17.7,16.0.
经表征,所制备的化合物结构正确。
实施例26、式Ⅶ-2所示化合物的制备
将66mg式Ⅳ-7所示化合物溶于5ml二氯甲烷中,加入80mg m-CPBA(纯度大于80%)和24mg NaHCO3。室温下搅拌12h。TLC检测反应完成后水洗,用二氯甲烷萃取。所得有机相采用无水硫酸钠干燥,然后再用旋转蒸发仪蒸干溶剂,所得产品采用硅胶柱(乙酸乙酯:石油醚=1:3,体积比)分离得到53mg式Ⅶ-2所示化合物。
1H-NMR(400MHz,CDCl3)δ(ppm)2.27(dd,J=12.2Hz,J=3.4Hz,1H),0.87(s,6H),0.74(s,3H);13C-NMR(100MHz,CDCl3)δ(ppm)175.0,85.9,71.5,71.2,69.8,49.3,48.4,44.4,44.1,43.4,40.5,36.6,36.3,33.1,32.4,32.3,31.8,29.4,29.3,28.5,26.9,25.6,24.9,22.0,21.6,21.2,20.1,19.1,19.0,16.5.
经表征,所制备的化合物结构正确。
下述实施例27-28所用的部分试验材料的来源如下:
本测试例用到的部分实验材料来源:
高纯度羊毛甾醇(L5768)、胆固醇(C3045)、二甲基亚砜(D8418)、多聚甲醛、Triton X-100和NP-40(Nonidet P40)购买自Sigma公司。
DAPI-Fluoromount-G荧光封片剂(0100-20)购买自SouthernBiotech(SBA)。
Anti-p62抗体(ab56416)购买自Abcam公司。
羊毛甾醇粗品购买自TCI(C0427),纯度>50.0%(GC)。
细胞培养基、抗生素、转染试剂LipofectamineTM 2000均购买自Invitrogen公司。
Cell counting kit-8(CK04-500)购买自日本同仁化学。
其他未注明具体来源的化学试剂均为分析纯。
晶状体蛋白αA-Y118D、αB-R120G、βB2-V187E、γC-G129C和γD-W43R突变体为本研究组克隆构建,具体过程如下:
将DNA分子1插入pEGFP-N1载体(Clontech)的XhoⅠ和BamHⅠ酶切位点之间,得到重组质粒1。DNA分子1:将序列表的序列1所示的DNA分子的第354位核苷酸由T突变为G(相应的第118位氨基酸由Y突变为D)得到的DNA分子。序列表的序列1所示的DNA分子为CRY-αA的开放阅读框。
将DNA分子2插入pEGFP-N1载体(Clontech)的XhoⅠ和HindⅢ酶切位点之间,得到重组质粒2。DNA分子2:将序列表的序列2所示的DNA分子的第360位核苷酸由A突变为G(相应的第120位氨基酸由R突变为G)得到的DNA分子。序列表的序列2所示的DNA分子为CRY-αB的开放阅读框。
将DNA分子3插入pEGFP-N1载体(Clontech)的XhoⅠ和HindⅢ酶切位点之间,得到重组质粒3。DNA分子3:将序列表的序列3所示的DNA分子的第562位核苷酸由T突变为A(相应的第187位氨基酸由V突变为E)得到的DNA分子。序列表的序列3所示的DNA分子为CRY-βB2的开放阅读框。
将DNA分子4插入pEGFP-N1载体(Clontech)的XhoⅠ和HindⅢ酶切位点之间,得到重组质粒4。DNA分子4:将序列表的序列4所示的DNA分子的第387位核苷酸由G突变为T(相应的第129位氨基酸由G突变为C)得到的DNA分子。序列表的序列3所示的DNA分子为CRY-γC的开放阅读框。
将DNA分子5插入pEGFP-N1载体(Clontech)的XhoⅠ和HindⅢ酶切位点之间,得到重组质粒5。DNA分子5:将序列表的序列5所示的DNA分子的第129位核苷酸由T突变为C(相应的第43位氨基酸由W突变为R)得到的DNA分子。序列表的序列3所示的DNA分子为CRY-γD的开放阅读框。
HeLa( CCL-2TM),人类晶状体上皮细胞HLE-B-3( CRL-11421TM)均购买自ATCC细胞库,培养条件按ATCC公司所建议的条件进行。
本发明测试例中未注明具体条件的实验方法,通常按照常规条件。
实施例27、本发明化合物对晶状体蛋白突变体在细胞内聚集的影响
本实施例用以下方法来测定本发明化合物对晶状体蛋白αA-Y118D、αB-R120G、βB2-V187E、γC-G129C和γD-W43R突变体在HeLa细胞内聚集情况的影响。
测试中以溶剂DMSO为空白对照,Lanosterol(羊毛甾醇)为阳性对照,Cholesterol(胆固醇)为阴性对照,并测试了现有化合物C29,作为本发明化合物的对照。具体方法如下:
(1)以晶状体蛋白αB-R120G突变体为研究模型研究本发明化合物的性能
晶状体蛋白αB-R120G突变体在细胞内发生错误折叠,形成显微镜下可见的聚集小体,如图1所示,其形成聚集小体的形态均一,聚集比例稳定,是研究化学药物缓解蛋白质错误折叠的理想模型。因此,本发明首先采用晶状体蛋白αB-R120G突变体为研究模型筛选有效的化合物。
具体实验方案如下:
将状态良好的HeLa细胞接种到预先已铺好细胞爬片的12孔板中,细胞密度40%~50%;培养24h左右,细胞密度80%左右,即可以进行细胞转染,具体操作按照Invitrogen公司的转染试剂LipofectamineTM 2000所建议方案进行;转染4h后,换成DMEM新鲜培养基继续培养16h,这时晶状体蛋白αB-R120G突变体在细胞内已大量表达,形成一定比例的聚集小体;将细胞培养基换做opti-MEM,并加入本发明化合物,至终浓度为4uM;培养4h后,换成DMEM新鲜培养基,再培养4h;进行免疫荧光标片的制备:PBS洗细胞爬片3遍,4%多聚甲醛室温固定细胞30分钟,PBS洗细胞爬片3遍,0.4%Triton X-100室温孵育细胞爬片15分钟,PBS洗细胞爬片3遍,4%山羊血清室温封闭40分钟,p62抗体室温孵育1小时,PBS洗细胞爬片3遍,相应来源的荧光二抗室温孵育40分钟,PBS洗细胞爬片3遍,用DAPI-Fluoromount-G荧光封片剂封片,室温避光放置1小时左右;应用Zeiss 710三通道显微镜进行观察,采取单盲统计细胞内的聚集情况,分析本发明化合物的药效。
所有样品设三组平行实验,并对实验进行了三次重复,通过上述方法统计分析本发明化合物对晶状体蛋白αB-R120G突变体在HeLa细胞内聚集情况,结果如表1中所示。
表1中数据显示,本发明化合物对对晶状体蛋白αB-R120G突变体在HeLa细胞内形成的聚集现象具有缓解作用,尤其是式Ⅲ-3所示化合物、式Ⅲ-6所示化合物式、Ⅲ-8所示化合物;式Ⅳ-1所示化合物、式Ⅳ-7所示化合物和式Ⅳ-12所示化合物对晶状体蛋白αB-R120G突变体在HeLa细胞内形成的聚集现象具有明显的缓解作用
表1本发明化合物对晶状体蛋白αB-R120G突变体在细胞内聚集影响的统计
(2)多种晶状体蛋白突变体中验证本发明化合物的有效性。
晶状体内的蛋白质突变后发生错误折叠,大多都会导致白内障疾病。因此,在多种晶状体蛋白突变体中验证本发明化合物的药效是否具有普遍效应对白内障疾病的治疗具有重要的意义。
除αB R120G突变体外,本发明选用了晶状体蛋白αA-Y118D、βB2-V187E、γC-G129C和γD-W43R突变体对初步筛选有效的药物做进一步验证。具体方案同药物筛选的过程。通过进一步的验证,结果表明,本发明化合物式Ⅲ-6和式Ⅳ-1在多种晶状体蛋白突变体形成聚集的细胞模型中都具有明显的效果,如图2所示。
(3)初步标定本发明物的药效
本本发明初步以晶状体蛋白αB-R120G突变体为研究模型,标定本发明式Ⅲ-6所示化合物的半效应浓度EC50值,如图3所示,结果显示式Ⅲ-6所示化合物的药效要比天然化合物羊毛甾醇有着数量级上的提升,白内障疾病的药物治疗具有巨大的潜力。
实施例29、本发明化合物细胞毒性的检测
安全有效的药物是本发明的研究原则,本发明将进一步检测本发明式Ⅲ-6所示化合物对细胞是否具有细胞毒性。测试设立1个对照组,转染空载体的HLE-B3细胞;5个实验组,转染晶状体蛋白αA-Y118D、αB-R120G、βB2-V187E、γC-G129C和γD-W43R突变体的HLE-B3细胞,模拟不同晶状体蛋白突变导致的白内障疾病。
用以下方法检测本发明化合物对不同细胞株的细胞毒性:
将状态良好的HLE-B3细胞接种到12孔板中,细胞密度40%~50%;培养24h左右,细胞密度80%左右,即可以进行细胞转染,分别转染晶状体蛋白αA-Y118D、αB-R120G、βB2-V187E、γC-G129C和γD-W43R突变体,及一个空载对照组。具体操作按照Invitrogen公司的转染试剂LipofectamineTM 2000所建议方案进行;转染4h后,换成DMEM新鲜培养基继续培养16h,这时晶状体蛋白突变体在细胞内已大量表达;用0.25%胰酶(EDTA)消化吹散后,接种到96孔板,每孔2000细胞,继续培养12小时;将细胞培养基换做opti-MEM,并加入本发明化合物1G,设定两个浓度梯度,终浓度为5uM或50nM,孵育12小时;加入10μL CKK-8,孵育1小时,450nm测吸收值。
所有样品设三组平行实验,并对实验进行了三次重复,通过上述方法统计分析本发明式Ⅲ-6所示化合物对正常晶状体上皮细胞以及转染各种晶
一种化合物及其在治疗白内障中的应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












动态评分
0.0