

IPC分类号 : C07J41/00,A61P9/00,A61P9/04,A61P9/06








专利摘要
本发明属于强心药物制备技术领域,公开了一种对Na+/K+?ATP酶α2亚型有选择性抑制作用的蟾毒灵核糖苷及其应用。所述的对Na+/K+?ATP酶α2亚型有选择性抑制作用的蟾毒灵核糖苷具有如式(一)所示的结构式:本发明首次合成了蟾毒灵核糖苷,该蟾毒灵核糖苷安全性高,对α1、α2亚型的选择性达到17.0,强心作用强,可制备成各种剂型,预示着良好的药用前景。且制备工艺简单,成本低,产率高。
权利要求
1.一种对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷,其特征在于具有如式(一)所示结构式:
2.一种根据权利要求1所述的对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷的制备方法,包括以下步骤:
(1)将蟾毒灵与氯铬酸吡啶盐酸盐以摩尔比1:2混合,溶于二氯甲烷中,室温下反应2.5h后,硅胶柱色谱分离,得到蟾毒灵3位氧化的中间体Bufalinone;
(2)将氧化产物Bufalinone与甲氧胺盐酸盐以摩尔比1:2.5混合,溶于甲醇中,加入吡啶,室温下反应2.5~3h,得到肟式中间体;
(3)在肟式中间体中加入3倍肟式中间体摩尔量的叔丁胺基甲硼烷盐酸盐复合物,然后用体积比为2:1的二氧六环/乙醇混合液溶解,冰水浴条件下反应3.5h,硅胶柱色谱分离,得到蟾毒灵3位α和β两种构型的苷元异构体;
(4)分别将步骤(3)得到的两种苷元异构体与L-(+)-核糖置于体积比为3:1的二甲基甲酰胺/乙醇混合液中反应48h,经高效液相分离得到蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷和蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷。
3.根据权利要求1所述的对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷在制备强心药物中的应用。
4.根据权利要求3所述的对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷在制备强心药物中的应用,其特征在于:所述的药物含有蟾毒灵核糖苷、其药用盐和其溶剂化物中的至少一种。
5.根据权利要求3所述的对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷在制备强心药物中的应用,其特征在于:所述的药物含有一种或多种药学上可接受的载体或赋形剂。
6.根据权利要求5所述的对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷在制备强心药物中的应用,其特征在于:所述的赋形剂包括稀释剂、湿润剂、润滑剂、填充剂和防腐剂中的至少一种。
7.根据权利要求6所述的对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷在制备强心药物中的应用,其特征在于:稀释剂为乳糖、淀粉、糊精、微晶纤维素和微粉硅胶中的至少一种。
8.根据权利要求6所述的对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷在制备强心药物中的应用,其特征在于:所述的润滑剂为水和50~85%不同浓度的乙醇。
9.根据权利要求3所述的对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷在制备强心药物中的应用,其特征在于:所述的药物采用本领域的常规方法制备成各种剂型,包括胶囊、丸剂、片剂、口服液、颗粒剂、酊剂的剂型及注射液。
说明书
技术领域
本发明属于强心药物制备技术领域,特别涉及一种对Na+/K+-ATP酶α2亚型有选择性抑制作用的蟾毒灵核糖苷及应用。
背景技术
心衰是严重的心血管系统疾病。据统计,每年我国的心衰患者约四百万。强心甾为常用的治疗药物。Na+/K+-ATP酶是强心甾的作用靶标。强心甾通过抑制Na+/K+-ATP酶,使细胞内Na+浓度上升,启动Na+-Ca2+交换,使细胞内Ca2+浓度上升,从而发挥强心作用。常用的强心甾类药物有地高辛、洋地黄毒苷、毛花苷丙、毒毛花苷K及福寿草总苷等,均为甲型强心甾。其中地高辛的应用已超过200年。这些药物在临床上用于治疗心力衰竭和某些心律失常,如心房纤颤及心动过速等(Gheorghiade,M.;Adams,K.F.;Colucci,W.S.Digoxininthemanagementofcardiovasculardisorders.Circulation,2004,109,2959-2964.)。但是甲型强心甾类药物的安全窗口小,一般治疗量已经接近于中毒剂量的60%,故甲型强心甾的中毒案例时有发生(Chan,K.E.;Lazarus,J.M.;Hakim,R.M.DigoxinassociateswithmortalityinESRD.J.Am.Soc.Nephrol,2010,21,1550-1555.)。虽然已有多个甲型强心甾类药物应用于临床,但其安全窗口小,故急需新型替代强心药物。
Na,K-ATPase含有α和β亚基,其中α亚基可分为α1、α2、α3、α4四个亚型,β亚基可分为β1、β2、β3三个亚型。α1亚型是广泛存在的,而其它亚型分布于特定的组织中。其中,α2亚型主要分布于肌肉中(骨骼肌、平滑肌和心脏)。在心肌细胞中α2亚型集中于肌浆网旁的T-小管中,而α1亚型均匀分布于肌浆网和T-小管中(Garty,H.,andKarlish,S.J.(2006)Annu.Rev.Physiol.68,431–459)。
强心甾通过抑制Na+/K+-ATP酶,使细胞内Na+浓度上升,启动Na+-Ca2+交换,使细胞内Ca2+浓度上升,从而发挥强心作用。但同时,强心甾的毒性源于对Na+/K+-ATP酶的过度抑制、Ca2+超载,并且肌浆网释放Ca2+,引发延迟后去极化和心率失常(Bers,D.M.(2008)Annu.Rev.Physiol.70,23–49)。国外学者采用三种转基因小鼠(对哇巴因不敏感的α1和敏感的α2,即α1R/Rα2S/S,对哇巴因敏感的α1和不敏感的α2,即α1S/Sα2R/R,和对哇巴因不敏感的α1和不敏感的α2,即α1R/Rα2R/R进行实验,发现α2亚型对于正性肌力作用起主导作用(Dostanic-Larson,I.,Lorenz,J.N.,VanHuysse,J.W.,Neumann,J.C.,Moseley,A.E.,andLingrel,J.B.(2006)Am.J.Physiol.Regul.Integr.Comp.Physiol.290,R524–R528),并且对于心肌T-小管中电流贡献最大(Despa,S.,andBers,D.M.(2007)Am.J.Physiol.CellPhysiol.293,C321–C327)。因此,对α2亚型有选择性抑制的强心甾,其在诱导正性肌力作用的同时,将最低限度地导致Ca2+超载,可作为较安全的强心药。
近年来,国内外学者一直在寻找对α2亚型有选择性抑制的强心甾。文献曾报道20余种强心甾对α1、α2亚型的选择性,发现地高辛有最高的选择性,其抑制常数Ki(α1)/Ki(α2)为3.96±0.93,而哇巴因的值为1.03±0.18。本课题组曾合成蟾毒灵-3β-N-甲氧基-N-β-D-葡萄糖苷,发现其Ki(α1)/Ki(α2)值为4.0,该值与地高辛相似。
与葡萄糖为六碳糖不同的是,核糖是一种五碳醛糖,一般常见的型态为D-核糖,是RNA的组成物之一,也是ATP及NADH等生化代谢所需分子的原料。D-核糖是核酸中的碳水化合物组分,以呋喃糖型广泛存在于植物和动物细胞中。并且,D-核糖的对映体L-核糖在自然界不存在,一般只能通过合成的方法得到[有机化学,2002年第22(3):153~158]。因此,L-核糖是非天然且不易得到的单糖,较少地用于药物合成。在蟾毒灵等蟾蜍二烯内酯的结构修饰中,核糖苷未见报道。
为了进一步提高选择性指数,我们首次尝试将L-核糖与蟾毒灵进行连接,意外发现蟾毒灵的核糖苷具有比葡萄糖苷更强的选择性。
发明内容
为了克服上述现有技术的缺点与不足,本发明的首要目的在于提供一种对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷。本发明的蟾毒灵核糖苷对α1、α2亚型的选择性达到17.0。
本发明另一目的在于提供上述蟾毒灵核糖苷的制备方法。
本发明再一目的在于提供上述蟾毒灵核糖苷在制备强心药物中的应用。
本发明的目的通过下述方案实现:
一种对Na+/K+-ATP酶α2亚型具有选择性抑制作用的蟾毒灵核糖苷,其具有如式(一)所示的结构:
所述的蟾毒灵核糖苷为氮苷结构,其中,苷元部分为蟾毒灵,糖基部分为L-(+)-核糖(L-(+)-Ribose),氮原子上有甲氧基取代。
一种上述蟾毒灵核糖苷的制备方法,包括以下步骤:
(1)将蟾毒灵与氯铬酸吡啶盐酸盐以摩尔比1:2混合,溶于二氯甲烷中,室温下反应2.5h后,硅胶柱色谱分离,得到蟾毒灵3位氧化的中间体Bufalinone;
(2)将氧化产物Bufalinone与甲氧胺盐酸盐以摩尔比1:2.5混合,溶于甲醇中,加入吡啶,室温下反应2.5~3h,得到肟式中间体;
(3)在肟式中间体中加入3倍肟式中间体摩尔量的叔丁胺基甲硼烷盐酸盐复合物,然后用体积比为2:1的二氧六环/乙醇混合液溶解,冰水浴条件下反应3.5h,硅胶柱色谱分离,得到蟾毒灵3位α和β两种构型的苷元异构体;
(4)分别将步骤(3)得到的两种苷元异构体与L-(+)-核糖置于体积比为3:1的二甲基甲酰胺/乙醇混合液中反应48h,经高校液相分离得到蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷和蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷。
制备方法参见CN103288911B,区别仅在于将步骤(4)中还原糖替换为L-(+)-核糖,本发明首次报道α和β-构型的苷元与L-(+)-核糖的反应。
上述的蟾毒灵核糖苷在制备强心药物中的应用。
所述的药物含有蟾毒灵核糖苷、其药用盐和其溶剂化物中的至少一种。
所述的药物含有一种或多种药学上可接受的载体或赋形剂。
所述的赋形剂可包括稀释剂、湿润剂、润滑剂、填充剂、防腐剂等。其中稀释剂可为乳糖、淀粉、糊精、微晶纤维素、微粉硅胶等,润湿剂可为水和50~85%不同浓度的乙醇。
所述的药物可以采用本领域的常规方法制备成各种剂型,如胶囊、丸剂、片剂、口服液、颗粒剂、酊剂等口服给药的剂型及注射液等口服以外的给药剂型,如针剂等。
本发明相对于现有技术,具有如下的优点及有益效果:
(1)本发明首次合成了蟾毒灵核糖苷。
(2)本发明揭示了蟾毒灵核糖苷发掘了的医疗用途,开拓了一个新的应用领域。
(3)本发明的蟾毒灵核糖苷安全性高,强心作用强,预示着良好的药用前景。
(4)本发明的蟾毒灵核糖苷的制备工艺简单,成本低,产率高。
附图说明
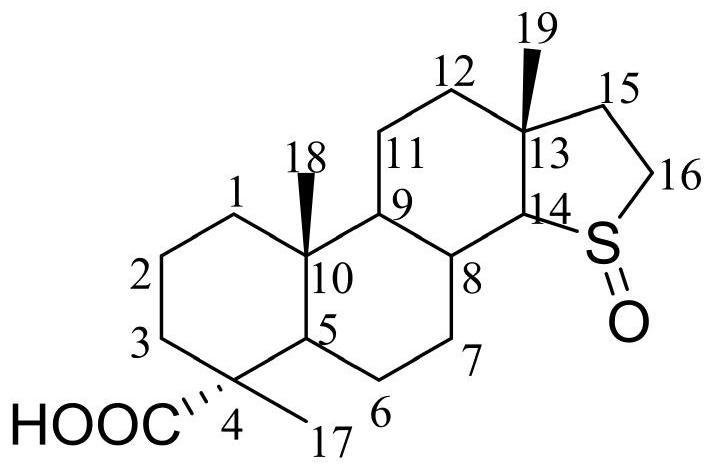
图1为蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)和蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷(化合物2)的化学结构式。
图2为蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)对斑马鱼心脏形态的影响,横坐标处的“1”代表化合物1。
图3为蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)的强心作用效果图。
具体实施方式
下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例中所用试剂均可从市场常规获得。
实施例1:蟾毒灵核糖苷的合成
(1)将蟾毒灵与氯铬酸吡啶盐酸盐以摩尔比1:2混合,溶于二氯甲烷中,室温下反应2.5h后,硅胶柱色谱分离,得到蟾毒灵3位氧化的中间体Bufalinone;
(2)将氧化产物Bufalinone与甲氧胺盐酸盐以摩尔比1:2.5混合,溶于甲醇中,加入吡啶,室温下反应2.5~3h,得到肟式中间体,收率较高(产率90%);
(3)在肟式中间体中加入3倍肟式中间体摩尔量的叔丁胺基甲硼烷盐酸盐复合物,然后用体积比为2:1的二氧六环/乙醇混合液溶解,冰水浴条件下反应3.5h,硅胶柱色谱分离,得到蟾毒灵3位α和β两种构型的苷元异构体,收率约60%,其中,中间体摩尔比约为α构型的苷元异构体:β构型的苷元异构体=2:1;
(4)分别将步骤(3)得到的两种苷元异构体与L-(+)-核糖(购自百灵威试剂公司)置于体积比为3:1的二甲基甲酰胺/乙醇混合液中反应48h,经高效液相分离得到蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)和蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷(化合物2)。
具体操作参见CN103288911B,区别仅在于将步骤(4)中的还原糖替换为L-(+)核糖。蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)和蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷(化合物2)的结构式见图1。
蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)的波谱数据:1HNMR(DMSO-d6,400MHz)δ7.93(dd,1H,J=9.6,2.5Hz),7.51(d,1H,J=2.5Hz),6.28(d,1H,J=9.6Hz),4.63(d,0.5H,J=3.3Hz,α-H1),4.20(d,0.5H,J=8.8Hz,β-H1),4.04(t,0.5H,α),4.02(t,0.5H,J=4.8Hz,β),3.72(m,0.5H,α),3.68(m,0.5H,β),3.63(dd,0.5H,J=10.8,6.0Hz,α),3.53(m,0.5H,β),3.19,(m,1H,2α),3.10(m,1H,2β),3.53(s,3H),3.10(m,1H),2.16,1.82(m,2H),2.12,1.39(m,2H),2.10,1.32(m,2H),2.07,1.60(m,2H),1.77,1.24(m,2H),1.79,1.18(m,2H),1.60,1.38(m,2H),2.46(m,1H),2.05(m,1H),1.93(m,1H),1.80(m,1H),1.61(m,1H),1.50(m,1H),0.88(s,3H),0.60(s,3H).13CNMR(DMSO-d6,100MHz)δ161.3,149.2,147.4,122.8,114.2,94.4(α-C1),85.8(β-C1),83.4,82.5(α),71.0(α),70.6(α),70.5(β),67.3(β),67.0(β),64.1(α),63.2,62.6(β),57.0,50.1,48.1,41.4,40.0,35.6,35.5,35.3,32.1,32.0,30.4,29.0,28.5,26.7,23.9,21.3,21.1,16.7.ESI-MSm/zC30H45NO8:548.3[M+H]+,570.3[M+Na]+,1117.4[2M+Na]+;HRMS(ESI)m/zC30H45NO8Na([M+Na]+)570.3044,cacl.570.3042.
根据质谱、核磁共振信息,鉴定产物为蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷,其化学式为C30H45NO8。
蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷(化合物2)的波谱数据:1HNMR(DMSO-d6,300MHz)δ7.93(dd,1H,J=9.8,2.3Hz),7.52(d,1H,J=2.3Hz),6.28(d,1H,J=9.8Hz),4.61(d,0.5H,J=3.8Hz,α-H1),4.35(t,0.5H,J=5.4Hz,β),4.22(d,0.5H,J=8.8Hz,β-H1),4.00(t,J=4.2Hz,0.5H,α),3.74(t,0.5H,J=4.8Hz,α),3.63(dd,0.5H,J=10.5,5.4Hz,α),3.53-3.51(m,1H,2β),3.37(m,1H,2α),3.45(m,1H,2β),3.48(s,3H),2.88(m,1H),2.07,1.61(m,2H),1.95,1.60(m,2H),1.78,1.37(m,2H),1.76,1.25(m,2H),1.73,1.34(m,2H),1.53,1.32(m,2H),1.33,1.07(m,2H),2.44(m,1H),1.58(m,1H),1.50(m,1H),1.28(m,1H),0.85(s,3H),0.58(s,3H).13CNMR(DMSO-d6,75MHz)δ161.4,149.2,147.4,122.8,114.2,95.4(α-C1),86.0(β-C1),83.4,82.8(α),71.4(β),71.0(α),71.0(α),67.2(β),67.0(β),64.2(α),63.4,62.6(β),61.7,50.1,48.1,41.5,41.3,40.0,35.8,35.0,34.6,32.1,29.8,28.5,27.1,25.3,23.4,21.3,20.9,16.7.ESI-MSm/zC30H45NO8:548.3[M+H]+,570.3[M+Na]+,1117.1[2M+Na]+;HRMS(ESI)m/zC30H45NO8Na([M+Na]+)570.3048,cacl.570.3042.
根据质谱、核磁共振信息,鉴定产物为,蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷,其化学式为C30H45NO8。
实施例2:蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)和蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷(化合物2)的Na+/K+-ATP酶α1、α2亚型的选择性。
测试方法参见文献(THEJOURNALOFBIOLOGICALCHEMISTRY2010.285,pp.19582–19592),对化合物1和2进行抑制常数(Ki)的测定,具体步骤如下:
(1)将活性测试溶液加入到48孔板中,每孔400μL,含有100mmol/LNaCl,20mmol/LKCl,1mmol/LMgCl2,1mmol/LEGTA,20mmol/LTris-HCl,pH7.4,和0.2μg纯化的Na+/K+-ATP酶α1或α2亚型(用酵母Pichiapastoris表达)
(2)加入不同浓度的化合物1和2后,将96孔板于37℃孵育15分钟。
(3)加入2mmol/LATP-Mg(美国Sigma公司)启动酶促反应,并在37℃孵育15分钟。
(4)将反应中释放的磷酸用BIOMOLGreen试剂(深圳欣博盛生物科技有限公司)(200μL)染色,根据620nm的吸光度来测定释放磷酸的量。
(5)根据方程进行拟合,求出抑制常数Ki值,该方程为V化合物/V0=Ki/([化合物]+Ki)+c(Kaleidagraph常数).其中,V0和V化合物分别指的是对照速率和加入特定化合物后的速率。
本实验测得蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)对Na+/K+-ATP酶α1的Ki为255nM,对α2亚型的Ki为15nM,对Ki(α1)/Ki(α2)亚型的选择性指数为17.0;蟾毒灵-3α-N-甲氧基-N-L-(+)核糖苷(2)对Na+/K+-ATP酶α1、α2亚型的Ki值均超过1μM,而相同条件下阳性药地高辛(digoxin)的IC50对α1、α2亚型的Ki值分别为245nM和41nM,Ki(α1)/Ki(α2)值为6.0。可见,化合物1对Na+/K+-ATP酶的抑制活性强于化合物2及阳性药地高辛,并且,化合物1对对α2亚型的选择性指数显著强于阳性药地高辛。
实施例3:蟾毒灵-3β-N-甲氧基-L-(+)核糖苷(化合物1)对斑马鱼的强心作用
本发明采用心脏带有绿色荧光(便于观察)的斑马鱼模型来研究其强心作用(Huang,C.C.;Monte,A.;Cook,J.M.;Kabir,M.S.;Peterson,K.P.ZebrafishHeartFailureModelsfortheEvaluationofChemicalProbesandDrugs.AssayDrugDev.Technol.2013,11(9-10),561-572.)。
采用Tg(cmlc2:GFP)转基因斑马鱼(由澳门大学提供),其受精卵由清晨雌雄鱼交配获得。将受精卵分成四组(溶媒对照组、模型组、给药组、阳性对照组)然后将各组受精卵放于培养液中,28℃孵化。24h后,向培养液中加入多柔比星(0.3μM),并继续培养24小时,造模),用数字化荧光倒置显微镜观察斑马鱼的心脏形态并测定心率以确定心衰模型构建成功。然后,向给药组培养液中加入不同浓度的化合物1,阳性对照组中加入地高辛(0.5μM),溶媒对照组中加入0.1%的二甲基亚砜DMSO,并继续培养48小时后,用数字化荧光倒置显微镜观察各化合物对受损心脏形态的恢复作用,并根据文献报道的方法(Moore,F.B.G.;HoseyM.;Bagatto,B.Cardiovascularsysteminlarvalzebrafishrespondstodevelopmentalhypoxiainafamilyspecificmanner.FrontiersinZoology,2006,3,4.)测定心率、每搏输出量(strokevolume)、心输出量(cardiacoutput)等心脏功能指标来评价化合物的强心活性,结果见图2。图2中的1指的是化合物1。
结果表明,用多柔比星造模后,心率加快,斑马鱼的每博输出量减少为溶媒对照组的65.6%,而心输出量降低为溶媒对照组的66.8%,表明造模成功。当加入浓度为0.1,0.2,0.5,1.0,2.0μmol/L的蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷后,心率基本不变,每博输出量显著增加,相对于模型组,增加量分别为14.2%,59.5%,90.8%,106.9%和126.1%(同样,斑马鱼的心输出量也显著增加,增加量分别为23.1%,49.2%,79.8%,102.1%和117.5%。在相同条件下,阳性药地高辛(0.5μmol/L)的每博输出量和心输出量的增加量分别为65.5%和69.2%。可见,本发明蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷的强心作用具有剂量依赖性,并且,在相同浓度(0.5μmol/L)下,蟾毒灵-3β-N-甲氧基-L-(+)核糖苷的强心作用强于地高辛。
实施例4:蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)的斑马鱼的心脏毒性
采用野生型AB系斑马鱼(由澳门大学提供),其受精卵由清晨雌雄鱼交配获得。将受精卵分成四组(溶媒对照组、模型组、给药组、阳性对照组),然后将各组受精卵放于培养液中,28℃孵化。24h后,向给药组培养液中加入化合物1样品(0.3μmol/L),模型组培养液中加入多柔比星(0.3μmol/L),阳性药组培养液中加入地高辛(0.3μmol/L),溶媒对照组中加入0.1%的二甲基亚砜DMSO。并继续培养48h。将斑马鱼胚胎用3%甲基纤维素固定并用3-氨基苯甲酸乙酯甲磺酸盐(0.16mg/mL)进行麻醉。最后,用SZX10体视显微镜观察和比较四组斑马鱼的形态,结果见图3。图3中的1指是化合物1。
结果表明:与对照组相似,化合物1不引起斑马鱼心脏形态的变化,而地高辛和多柔比星组的外形均表现为脊柱弯曲。因此本发明蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(1)的整体毒性显著低于阳性药地高辛(digoxin)和多柔比星(doxorubicin,抗肿瘤药)。
实施例5:蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)在制备强心药中的应用
普通片剂的制备
取蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)50g、淀粉190g混合均匀,用70%乙醇制成软材,制粒,烘干。加入10g硬脂酸镁混合均匀,压片,制成2000片,包薄膜衣,每片含蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷25mg。
实施例6:蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)在制备强心药物总的应用
缓释片的制备
将蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)25g,用处方量85%乙醇溶液溶解,加辅料乳糖120g、羟丙基甲基纤维素(HPMC)110g、预胶化淀粉60g,混合均匀;加入85%乙醇溶液混合制成软材,制粒,过(18~35)目筛后干燥,将干颗粒过(18~35)目筛网整粒;称取硬脂酸镁10g,加入干颗粒中,混合均匀,浅弧形冲压片(1000片),即得蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)缓释片的素片,每片含蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷25mg。
实施例7:蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)在制备强心药物总的应用
口腔崩解片的制备
取蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)200g、微晶纤维素600g,交联聚维酮300g、甘露醇160g、硬脂酸镁170g、阿斯巴甜160g、柠檬酸10g,混合均匀,过80目筛,采用直接压片法制备片重为0.20g的口腔崩解片(8000片),每片含蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷25mg。
实施例8:蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)在制备强心药物总的应用
胶囊剂的制备
取蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)50g、乳糖200g、淀粉130g、硬脂酸镁20g,加65%乙醇制成软材,干燥、过筛、制粒,装入胶囊,制成2000粒。每粒胶囊含蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)25mg。
实施例9:蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)在制备强心药物总的应用
注射液的制备
蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)100g,丙二醇5000g,研磨,再加适量注射用水稀释,混匀,然后加入重量百分比为0.9%的氯化钠,溶解后再加入注射用水至100L,调pH值7.0~7.4,滤过,灌封,灭菌,即得1万支注射用针剂,每支含蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)10mg。
实施例10:蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)在制备强心药物总的应用
脂质体纳米粒的制备
蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)100g,大豆卵磷脂50g,溶于2500mL乙醇中,另取硬脂酸20g和大豆卵磷脂脂50g溶于2500mL环己烷中,混合搅拌均匀。于37℃恒温水浴中减压旋转蒸发除去有机溶剂,使药物及辅料在烧瓶壁形成均匀脂质薄膜,于真空干燥器中放置过夜,除尽有机溶剂;另取聚乙二醇单硬脂酸酯375g,搅拌溶解在17.5L水中,加入上述薄膜中,超声30min,定溶至25L,得淡黄色透明溶液。将此溶液冷冻干燥可得冻干粉。用球磨机研磨24小时,制得粒径均匀的纳米粒,混匀并分装。每袋含蟾毒灵-3β-N-甲氧基-N-L-(+)核糖苷(化合物1)10mg。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
对Na+/K+-ATP酶α2亚型有选择性抑制作用的蟾毒灵核糖苷及应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0