









专利摘要
本发明公开了一种纳米全硅beta分子筛及其制备方法,制备方法包括以下步骤:(1)将硅源、含氟和/或钠的矿化剂、模板剂和水混合均匀,得到碱与硅源的摩尔比为OH?:SiO2=0.21?4的反应混合物;(2)将步骤(1)得到的反应混合物转移至耐压的密闭容器中,并在60?115℃的温度和自生压力下老化0.05?30天,得到老化产物;(3)将步骤(2)得到的老化产物升温至130?230℃并在自生压力下晶化0.5?30天,得到晶化产物;其中所述晶化的温度比所述老化的温度高至少15℃;(4)回收步骤(3)得到的晶化产物。本技术方案得到的纳米全硅beta分子筛具有片状形貌,其长宽尺寸不大于490纳米,厚度不大于150纳米,29SiNMR的表征结果中Q4/Q3不小于25,比表面积SBET大于430m2/g。
权利要求
1.一种纳米全硅beta分子筛,其特征在于:该分子筛具有片状形貌,其长宽尺寸不大于490纳米,厚度不大于150纳米。
2.根据权利要求1的全硅beta分子筛,该分子筛的29SiNMR表征结果中Q4/Q3不小于25。
3.根据权利要求1的全硅beta分子筛,该分子筛的比表面积SBET大于430m2/g。
4.一种纳米全硅beta分子筛的合成方法,包括以下步骤:
(1)将硅源、含氟和/或钠的矿化剂、模板剂、水以及任选的碱源混合均匀,得到摩尔配比为OH-:SiO2:A:R:H2O=(0.21-4):1:(0.1-5):(0.4-5):(2-50)的反应混合物,其中A代表反应混合物中矿化剂的摩尔数,R代表反应混合物中模板剂的摩尔数;
(2)将步骤(1)得到的反应混合物转移至耐压的密闭容器中,并在60-115℃的温度和自生压力下老化0.05-30天,得到老化产物;
(3)将步骤(2)得到的老化产物在耐压的密闭容器中升温至130-230℃并在自生压力下晶化0.5-30天,得到晶化产物;
(4)回收步骤(3)得到的晶化产物。
5.根据权利要求4所述的方法,其中,步骤(1)中所述的反应混合物的摩尔配比为OH-:SiO2:A:R:H2O=(0.25-3):1:(0.2-2.5):(0.45-3):(3-30)。
6.根据权利要求4所述的方法,其中,步骤(1)中所述的硅源为选自硅酯、固体硅胶、白炭黑和硅溶胶中的至少一种。
7.根据权利要求4所述的方法,其中,步骤(1)中所述的矿化剂为选自氢氟酸、氟化钠、氟化钾、氟化铵、氟硅酸、四乙基氟化铵、氟硅酸铵、氯化钠、氟化钠、溴化钠、碘化钠、氢氧化钠、碳酸钠和碳酸氢钠中的至少一种。
8.根据权利要求4所述的方法,其中,步骤(1)中所述的矿化剂为选自氢氟酸、氟硅酸、氟化钠和氯化钠中的至少一种。
9.根据权利要求4所述的方法,其中,步骤(1)中所述的模板剂为选自四乙基氢氧化铵、四乙基氟化铵、四乙基氯化铵、四乙基溴化铵、四乙基碘化铵和三乙胺中的至少一种。
10.根据权利要求4所述的方法,其中,步骤(1)中所述的模板剂为选自四乙基氢氧化铵、四乙基氟化铵和三乙胺中的至少一种。
11.根据权利要求4所述的方法,其中,步骤(1)中所述的碱源为氢氧化钠、氨水、氢氧化锂、氢氧化铷、碳酸钠、碳酸氢钠和碳酸锂中的至少一种。
12.根据权利要求4所述的方法,其中,步骤(1)中所述的碱源为氢氧化钠。
13.根据权利要求4所述的方法,其中,步骤(1)中所述的将硅源、含氟和/或钠的矿化剂、模板剂、水以及任选的碱源混合均匀是先将硅源、模板剂和水以及任选的碱源在20-100℃的温度范围内混合均匀后,再加入矿化剂混合均匀。
14.根据权利要求4所述的方法,其中,步骤(2)中所述老化的温度为80-110℃,老化的时间为0.1-15天。
15.根据权利要求4所述的方法,其中,步骤(3)中所述晶化的温度为135-170℃,晶化的时间为1-20天。
16.根据权利要求4所述的方法,其中,该方法还包括步骤(5):将步骤(4)回收的晶化产物进行焙烧处理。
17.根据权利要求16所述的方法,其中,步骤(5)中所述焙烧处理的条件是:焙烧温度为400-800℃,焙烧时间为1-16小时。
说明书
技术领域
本发明涉及一种纳米全硅beta分子筛及其制备方法。
背景技术
β分子筛是R.Wadlinger等在1967年开发的具有BEA结构的分子筛,它的出现是高硅分子筛研究的标志。β分子筛是由几种结构不同但紧密相关的多形体组成的堆垛层错共生,具有三维十二元环孔道结构。其中,[100]和[010]方向的孔道为直孔道,其孔径都约为0.66×0.67纳米;[001]方向的孔道则是由[100]和[010]两个方向的直孔道交叉形成的正弦形结构,其孔径约为0.55×0.55纳米。
由于β分子筛具有高骨架硅铝比及特殊的三维十二元环孔道结构,因此具有热稳定性好、憎水性强、大分子反应物和产物在分子筛中的扩散性能好等优点,被广泛用于催化裂化、烷基化和异构化等反应中,是一种重要的石油化工催化剂。
由于β分子筛具有优异的催化性能和广阔的应用领域,因此其合成与改性一直是研究热点之一,而全硅β分子筛的合成又是其中的一个重要方面。根据文献报道,全硅β分子筛的水热合成主要包括以下四个方面:(1)使用新模板剂合成;(2)中性含氟体系合成;(3)碱性条件合成;(4)酸性条件合成。
通常条件下,β分子筛的水热合成是以四乙基铵盐或四乙基氢氧化铵为模板剂,而在全硅β分子筛的合成研究中,J.vanderWaal(JChemSoc,ChemCommun,1994,10:1241-1242.)等报道了以二苯基二甲基氢氧化铵为模板剂制备全硅β分子筛,但此法需以脱硼的B-β分子筛为晶种。US5554356以4,4ˊ-三亚甲基双(N-苯基哌啶)或其衍生物为模板剂合成全硅beta分子筛。K.Tsuji和M.Davis(MicroporouMater,1997,11:53-64.)则以4,4ˊ-三亚甲基双(N-甲基-N-苯基哌啶)或4,4ˊ-三亚甲基双(N-甲基-N-环己基甲基哌啶)为模板剂合成了全硅β分子筛。
中性含氟体系合成全硅β分子筛的研究中,M.Camblor等(ChemCommun,1996,20:2365-2366.)首先报道以正硅酸乙酯为硅源、HF为氟源,在合成体系的物料摩尔配比为SiO2:TEAOH:HF:H2O=1:0.54:0.54:7.25、晶化温度为413K以及搅拌条件下进行全硅β分子筛的合成。结果表明,此条件下制得的全硅β分子筛具有削角的八面立方结构,其颗粒尺寸在0.5~5.0微米之间、相对结晶度可达100%;此外,分子筛中的多形体A与多形体B的比例接近于1,这大于常规β分子筛的相应值0.6。而D.Serrano(MicroporousMesoporousMater,2001,46:35-46.)等对中性含氟体系条件下全硅β分子筛的合成机理的研究表明,β分子筛是由固相硅胶直接转化形成的,且其尺寸较大,初始粒径可达7微米,晶化完成后更是可达到14微米。
虽然碱性条件下难以合成出全硅β分子筛,但在中性含氟体系合成的基础上,O.Larlus等(ChemMater,2005,17:881-886.)报道了碱性条件下控制全硅β分子筛形貌的方法,但此法需在中性含氟体系下进行预晶化,且晶化产物的尺寸仍然大于10微米。此外,O.Larlus等(MicroporousMesoporousMater,2006,93:55-61.)还以热解硅胶为硅源,以双四乙基氟硅酸(TEA2SiF6)为硅源与氟源,并在OH-:SiO2不大于0.2的碱性条件下制备了全硅β分子筛,但此法制备的全硅β分子筛的粒径仍大于10微米。
EP2236461则报道了酸性条件下全硅β分子筛的合成方法,在F-:SiO2=0.55~2.0、TEA+:SiO2=0.5~1.0、H2O:SiO2=2.0~4.0、F-:TEA+=1.1~2.0的物料摩尔配比范围内,以硅胶、白炭黑、正硅酸乙酯、无定形二氧化硅或酸性硅溶胶为硅源、以HF为氟源,物料混合均匀后,在40~80℃内老化一定时间,随后在300~600转/分钟、40~180℃等条件下晶化1~4天,即可制得颗粒大小在0.5~5微米范围内的全硅β分子筛。
虽然使用新模板剂、在中性、碱性或酸性含氟体系中都可以制备出全硅β分子筛。但使用新模板剂时,模板剂的合成复杂、成本较高,因此,新模板剂法难以工业化推广;中性含氟体系下制备全硅β分子筛的方法则存在模硅比高、合成周期长、晶化产物的颗粒尺寸大等问题;酸性条件下制备全硅β分子筛的方法具有物料范围广、晶化速度快等优点,但酸性条件下,HF对设备的腐蚀较为严重,且考虑到HF的毒性,此法亦不宜推广使用;而碱性含氟体系制备的全硅β分子筛存在颗粒大的缺点,而且此法的晶化产物颗粒大小分布不很均匀。
全硅β分子筛的颗粒尺寸越小,其催化性能越优越;但就本发明人所知,现有技术中尚未开发出平均粒径小于500纳米的全硅β分子筛产品,因此需要开发一种成本较低、环境友好且能合成颗粒尺寸小且分布均匀的全硅β分子筛的方法。
发明内容
本发明的目的是提供一种纳米全硅beta分子筛及其合成方法,该纳米全硅beta分子筛是在碱性条件下合成,且具有颗粒尺寸小、分布均匀、骨架缺陷少的特点。
为了实现上述目的,本发明提供一种纳米全硅beta分子筛,其特征在于:该分子筛具有片状形貌,其长宽尺寸不大于490纳米,厚度不大于150纳米,29SiNMR的表征结果中Q4/Q3不小于25,比表面积SBET大于430m2/g。
另一方面,本发明还提供了一种纳米全硅beta分子筛的合成方法,包括以下步骤:
(1)将硅源、含氟和/或钠的矿化剂、模板剂、水以及任选的碱源混合均匀,得到摩尔配比为OH-:SiO2:A:R:H2O=(0.21-4):1:(0.1-5):(0.4-5):(2-50)的反应混合物,其中A代表反应混合物中矿化剂的摩尔数,R代表反应混合物中模板剂的摩尔数;
(2)将步骤(1)得到的反应混合物转移至耐压的密闭容器中,并在60-115℃的温度和自生压力下老化0.05-30天,得到老化产物;
(3)在耐压的密闭容器中,将步骤(2)得到的老化产物升温至130-230℃并自生压力下晶化0.5-30天,得到晶化产物;
(4)回收步骤(3)得到的晶化产物。
本发明提供的纳米全硅beta分子筛的合成方法属于碱性条件下的合成方法,通过提高晶化前的反应混合物的碱度,同时采用分步晶化法,得到的产物具有片状形貌;颗粒尺寸小,长宽尺寸不大于490纳米,厚度不大于150纳米;晶型完整,骨架缺陷少;比表面积大,SBET均大于430m2/g,有利于物质扩散。
本发明的其他特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
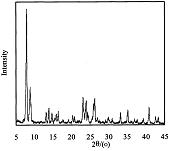
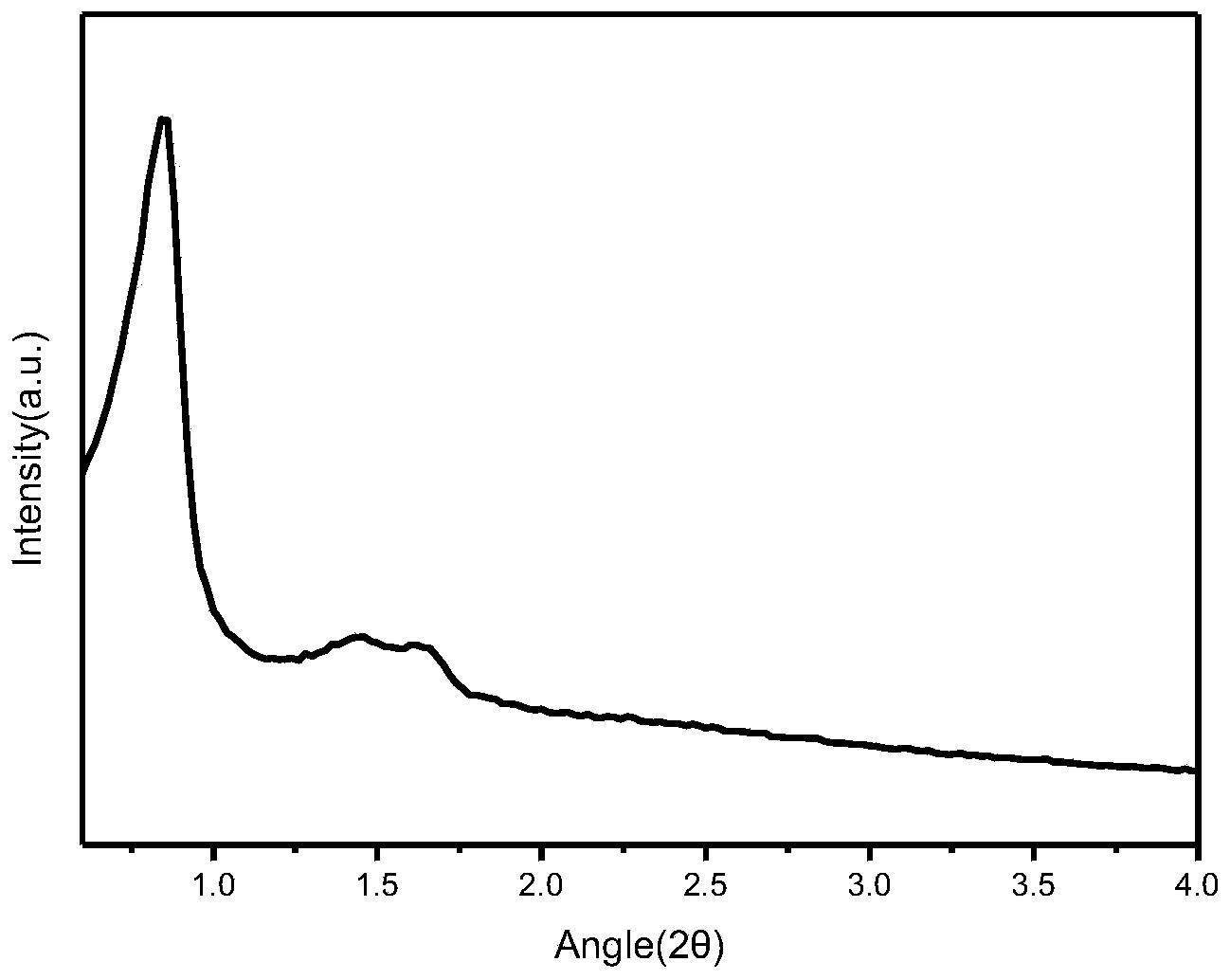
图1是按照本发明中合成beta分子筛的方法(实施例1)得到的beta分子筛的X射线衍射(XRD)的晶相图。
图2是按照本发明中合成beta分子筛的方法(实施例1)得到的beta分子筛的扫描电子显微镜(SEM)的形貌结果。
图3是按照本发明中合成beta分子筛的方法(实施例1)得到的beta分子筛的29SiNMR表征结果图。
图4是按照本发明中合成beta分子筛的方法(实施例4)得到的beta分子筛的扫描电子显微镜(SEM)的形貌结果。
图5是按照本发明中合成beta分子筛的方法(实施例4)得到的beta分子筛的29SiNMR表征结果图。
图6是按照现有技术(ChemCommun,1996,20:2365-2366.)在中性条件下合成全硅beta分子筛的方法(对比例1)得到的beta分子筛的X射线衍射(XRD)的晶相图。
图7是按照现有技术(ChemCommun,1996,20:2365-2366.)在中性条件下合成全硅beta分子筛的方法(对比例1)得到的beta分子筛的扫描电子显微镜(SEM)的形貌结果。
图8是按照在碱性条件下(MicroporousMesoporousMater,2006,93:55-61.)合成全硅beta分子筛的方法(对比例2)得到的beta分子筛的X射线衍射(XRD)的晶相图。
图9是按照在碱性条件下(MicroporousMesoporousMater,2006,93:55-61.)合成全硅beta分子筛的方法(对比例2)得到的beta分子筛的扫描电子显微镜(SEM)的形貌结果。
图10是按照在碱性条件下(MicroporousMesoporousMater,2006,93:55-61.)合成全硅beta分子筛的方法(对比例2)得到的beta分子筛的29SiNMR表征结果图。
图11是按照在酸性条件下(EP2236461)合成全硅beta分子筛的方法(对比例3)得到的beta分子筛的X射线衍射(XRD)的晶相图。
图12是按照在酸性条件下(EP2236461)合成全硅beta分子筛的方法(对比例3)得到的beta分子筛的扫描电子显微镜(SEM)的形貌结果。
具体实施方式
以下结合附图对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
本发明提供一种纳米全硅beta分子筛,其特征在于:该分子筛具有片状形貌,其长宽尺寸不大于490纳米,厚度不大于150纳米,29SiNMR的表征结果中Q4/Q3不小于25,比表面积SBET大于430m2/g。
根据本发明,所述的29SiNMR中的Q4信号是指分子筛中Si-(O-Si)4结构所产生的共振峰,即硅原子通过硅氧键与四个硅原子相连接所组成的结构产生的共振峰;Q3信号是指分子筛中HO-Si-(O-Si)3结构所产生的共振峰,即硅原子通过硅氧键与三个硅原子相连接并与一个羟基相连所组成的结构产生的共振峰。29SiNMR的表征结果说明,全硅beta分子筛只有强的Q4信号,而几乎没有Q3的信号,这说明此法制备的全硅beta分子筛几乎没有骨架缺陷。
根据本发明,所述的29SiNMR是使用VarianINOVA300型核磁共振波谱仪测定,测试条件为:59.588MHz,魔角转速为3kHz;对29SiNMR共振峰谱图进行分峰拟合后采用积分法计算各峰面积,Q4与Q3峰面积之比即为Q4/Q3值。
根据本发明,所述的比表面积是使用Micromeritics公司的ASAP2405J静态氮吸附仪在液氮温度(77.4K)下测得样品的静态N2吸脱附曲线后,对P/P0=0.05~0.35范围内的吸附曲线进行BET拟合得到。
另一方面,本发明还提供一种纳米全硅beta分子筛的合成方法,包括以下步骤:
(1)将硅源、含氟和/或钠的矿化剂、模板剂、水以及任选的碱源混合均匀,得到摩尔配比为OH-:SiO2:A:R:H2O=(0.21-4):1:(0.1-5):(0.4-5):(2-50)的反应混合物,其中A代表反应混合物中矿化剂的摩尔数,R代表反应混合物中模板剂的摩尔数;
(2)将步骤(1)得到的反应混合物转移至耐压的密闭容器中,在60-115℃的温度和自生压力下老化0.05-30天,得到老化产物;
(3)在耐压的密闭容器中,将步骤(2)得到的老化产物升温至130-230℃并在自生压力下晶化0.5-30天,得到晶化产物;
(4)回收步骤(3)得到的晶化产物。
根据本发明,优选的是,所述的步骤(1)中得到的所述反应混合物的摩尔比为OH-:SiO2:A:R:H2O=(0.25-3):1:(0.2-2.5):(0.45-3):(3-30);进一步优选为OH-:SiO2:A:R:H2O=(0.3-2.5):1:(0.25-1.5):(0.5-2):(4-10)。
根据本发明,所述的步骤(1)中的硅源、矿化剂、模板剂和水以及任选的碱源可以按照常规方法混合均匀,即制得所述反应混合物。
本发明的一种优选实施方式为:在步骤(1)中,可以先将硅源、模板剂、水以及任选的碱源在20-100℃的温度范围内混合均匀后,再加入矿化剂并混合均匀;进一步优化为先将硅源、模板剂和水以及任选的碱源在30-90℃的温度范围内混合均匀后,再加入矿化剂并混合均匀。
根据本发明,所述的步骤(1)中的反应混合物中的OH-既可以是来源于所述模板剂或者所述硅源中存在的OH-,也可以是来源于另外加入的所述碱源中的OH-;术语“任选的碱源”,是指当所加入的所述模板剂或者所述硅源中存在的OH-的量满足所述反应混合物的摩尔配比要求时,不需再另外加入碱源;而当OH-的量不能满足所述反应混合物的摩尔配比要求时,再另外加入所述碱源。所使用的所述碱源可以是本领域技术人员所熟知的合成beta分子筛时所常用的任意碱源,本发明对其没有特别的限制,例如该碱源可以是氢氧化钠、氨水、氢氧化锂、氢氧化铷、碳酸钠、碳酸氢钠和碳酸锂中的至少一种;优选的,步骤(1)中使用的碱源是氢氧化钠。
根据本发明,所述的步骤(1)中的硅源可以是本领域技术人员所熟知的合成全硅beta分子筛所常用的硅源,本发明对其没有特别的限制,例如该硅源可以是硅酯(有机硅酸酯)、固体硅胶、白炭黑和硅溶胶中的至少一种,其中的硅溶胶优选为碱性硅溶胶,碱性硅溶胶中的碱同时也可作为至少一部分的OH-源;为了避免硅源中的杂原子如硼或铝等三价杂原子对全硅β分子筛的晶化可能产生的影响,步骤(1)中所述的硅源优选为二氧化硅含量高而杂质含量少的硅酯、硅胶和白炭黑中的至少一种;进一步优选为硅酯和白炭黑中的至少一种,其中,所说的硅酯的通式为:
式中,R1、R2、R3和R4各自为C1-C4的烷基,包括C1-C4的直链烷基和C3-C4的支链烷基,如:R1、R2、R3和R4各自可以为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,其中优选的是R1、R2、R3和R4均为甲基或乙基。
根据本发明,所述的步骤(1)中使用的矿化剂可以是含卤素离子的化合物和含碱金属离子的化合物中的至少一种;如可以为含氟化合物与含钠化合物的混合物;其中,含氟化合物可以为选自氢氟酸、氟化钠、氟化钾(KF)、氟化铵、氟硅酸、四乙基氟化铵和氟硅酸铵中的至少一种;含钠化合物可以为选自氯化钠、氟化钠、溴化钠、碘化钠、氢氧化钠、碳酸钠和碳酸氢钠中的至少一种;优选的为选自氢氟酸、氟硅酸、氟化钠和氯化钠中的至少一种。
根据本发明,所述的步骤(1)中使用的模板剂可以是选自四乙基氢氧化铵、四乙基氟化铵、四乙基氯化铵、四乙基溴化铵、四乙基碘化铵和三乙胺中的至少一种,优选为四乙基氢氧化铵、四乙基氟化铵和三乙胺中的至少一种。
根据本发明,所述的步骤(1)中的水可以为合成分子筛时常用的水,为了避免杂原子的引入,本发明中优选为去离子水。
根据本发明,所述的步骤(2)中的老化条件优选为:老化温度为80-110℃,老化时间为0.1-15天。
根据本发明,所述的步骤(3)中的晶化条件优化为:晶化温度是135-170℃,晶化时间是1-20天。
根据本发明,所述的步骤(3)中的晶化可以是在静态条件下或动态搅拌条件下进行;为保证晶化体系均匀混合并获得均匀的晶化产物,晶化过程优化为在动态搅拌条件下进行;进一步优化为在300-800r/min的搅拌速度下进行动态晶化。
本发明提供的纳米全硅β分子筛的合成方法是在碱性条件下进行的,步骤(3)中所述的全硅β分子筛晶化完后,反应体系仍为碱性,其pH>9,进一步优化为pH>10。
根据本发明,所述的步骤(4)中的回收方法可以为常规回收法,如可以将步骤(3)得到的晶化产物经过滤、洗涤、干燥后得到干燥晶化产物;干燥温度可以为60-180℃,干燥时间可以为0.5-24小时,进一步优选为:干燥温度可以为90-130℃,干燥时间可以为2-12小时。
根据本发明,该合成方法还包括下列步骤(5):将步骤(4)回收的晶化产物进行焙烧处理,以脱除分子筛孔道中的模板剂。
根据本发明,所述的步骤(5)中所述焙烧处理的条件可以是:焙烧温度为400-800℃,焙烧时间为1-16小时。
以下将通过具体实施例对本发明进行详细描述。在以下各实施例及对比例中,X射线衍射(XRD)的晶相图是用PhilipsPanalyticalX'pert测定得到,测试条件为:Cu靶,Kα辐射,Ni滤波片,超能探测器,管电压30KV,管电流40mA;扫描电子显微镜(SEM)的形貌图是用Hitachi4800测定得到,加速电压为20KV,环境扫描;29SiNMR的表征图是用VarianINOVA300型核磁共振波谱仪测定,测试条件为:59.588MHz,魔角转速为3kHz;SBET结果是用Micromeritics公司的ASAP2405J静态氮吸附仪在液氮温度(77.4K)下测定。
实施例1
按SiO2:TEAOH=1:0.54的摩尔配比,再加入一定量的水,在600r/min的搅拌条件下,将正硅酸乙酯、去离子水以及四乙基氢氧化铵混合,并于70℃的温度下水解10h,得到澄清的正硅酸乙酯的水解溶液。在1000r/min的搅拌条件下,按照矿化剂:SiO2=0.5:1的摩尔配比,在正硅酸乙酯水解溶液中加入氟化钠和氢氟酸,搅拌均匀后得到H2O:OH-:SiO2=7.25:0.5:1的混合物。
将该混合物转移至密闭的耐压容器中,在400r/min的搅拌条件下,将晶化体系升温至110℃,并在自生压力下老化1天;随后继续在搅拌条件下将晶化体系升温至140℃,在自生压力下恒温10天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在110℃的温度下干燥6h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在550℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为4h,得到尺寸不大于490nm的全硅β分子筛,其X射线衍射(XRD)的晶相图如图1所示,扫描电子显微镜(SEM)的形貌结果如图2所示,29SiNMR的结果如图3所示。
实施例2
按SiO2:TEACl:LiOH:NaOH=1:0.8:0.7:0.1的摩尔配比,再加入一定量的水,在300r/min的搅拌条件下,将硅溶胶、去离子水、氢氧化钠、氢氧化锂以及四乙基氯化铵混合,并于30℃的温度下水解20h,得到硅溶胶的水解溶液。在100r/min的搅拌条件下,按照矿化剂:SiO2=0.2:1的摩尔配比,在硅溶胶水解溶液中加入氟硅酸,搅拌均匀后得到H2O:OH-:SiO2=10:0.73:1的混合物。
将该混合物转移至密闭的耐压容器中,在800r/min的搅拌条件下,将晶化体系升温至100℃,并在自生压力下老化5天;随后继续在搅拌条件下将晶化体系升温至150℃,在自生压力下恒温7天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在130℃的温度下干燥4h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在550℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为6h,得到尺寸不大于490nm的全硅β分子筛。
实施例3
按SiO2:TEABr:TEAOH:NaOH=1:0.4:0.6:0.2的摩尔配比,再加入一定量的水,在200r/min的搅拌条件下,将白炭黑、去离子水、四乙基氢氧化铵、氢氧化钠以及四乙基溴化铵混合,并于50℃的温度下水解12h,得到澄清的白炭黑的水解溶液。然后在300r/min的搅拌条件下,按照矿化剂:SiO2=0.25:1的摩尔配比,在白炭黑水解溶液中加入氟化钠,搅拌均匀后得到H2O:OH-:SiO2=17:0.8:1的混合物。
将该混合物转移至密闭的耐压容器中,在200r/min的搅拌条件下,将晶化体系升温至115℃,并在自生压力下老化0.05天;随后继续在搅拌条件下将晶化体系升温至135℃,在自生压力下恒温15天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在150℃的温度下干燥2h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在650℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为8h,得到尺寸不大于490nm的全硅β分子筛。
实施例4
按SiO2:TEAOH:NaOH=1:2.0:0.1的摩尔配比,再加入一定量的水,在700r/min的搅拌条件下,将硅胶、去离子水、氢氧化钠以及四乙基氢氧化铵混合,并于90℃的温度下水解6h,得到澄清的硅胶的水解溶液。然后在100r/min的搅拌条件下,按照矿化剂:SiO2=0.1:1的摩尔配比,在硅胶水解溶液中加入氟化铵,搅拌均匀后得到H2O:OH-:SiO2=20:2:1的混合物。
将该混合物转移至密闭的耐压容器中,在600r/min的搅拌条件下,将晶化体系升温至110℃,并在自生压力下老化0.1天;随后继续在搅拌条件下将晶化体系升温至170℃,在自生压力下恒温1天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在90℃的温度下干燥12h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在450℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为12h,得到尺寸不大于490nm的全硅β分子筛,扫描电子显微镜(SEM)的形貌结果如图4所示,29SiNMR的结果如图5所示。
实施例5
按SiO2:TEAF:TEAOH:LiOH=1:0.5:2.5:0.5的摩尔配比,再加入一定量的水,在600r/min的搅拌条件下,将白炭黑、氢氧化锂、去离子水、四乙基氟化铵以及四乙基氢氧化铵混合,并于100℃的温度下水解4h,得到澄清的白炭黑的水解溶液。然后在400r/min的搅拌条件下,按照矿化剂:SiO2=0.8:1的摩尔配比,在白炭黑水解溶液中加入氟化钠,搅拌均匀后得到H2O:OH-:SiO2=25:3:1的混合物。
将该混合物转移至密闭的耐压容器中,在800r/min的搅拌条件下,将晶化体系升温至70℃,并在自生压力下老化0.5天;随后继续在搅拌条件下将晶化体系升温至230℃,在自生压力下恒温0.5天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在60℃的温度下干燥24h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在400℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为16h,得到尺寸不大于490nm的全硅β分子筛。
实施例6
按SiO2:TEAOH:TEA:RuOH=1:4:1:0.5的摩尔配比,再加入一定量的水,在200r/min的搅拌条件下,将正硅酸乙酯、去离子水、三乙胺、氢氧化铷以及四乙基氢氧化铵混合,并于80℃的温度下水解8h,得到澄清的正硅酸乙酯的水解溶液。然后在400r/min的搅拌条件下,按照矿化剂:SiO2=1:1的摩尔配比,在正硅酸乙酯水解溶液中加入氟化钠和氟硅酸,搅拌均匀后得到H2O:OH-:SiO2=50:4:1的混合物。
将该混合物转移至密闭的耐压容器中,在500r/min的搅拌条件下,将晶化体系升温至60℃,并在自生压力下老化30天;随后继续在搅拌条件下将晶化体系升温至130℃,在自生压力下恒温20天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在120℃的温度下干燥4h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在500℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为5h,得到尺寸不大于490nm的全硅β分子筛。
实施例7
按SiO2:TEAOH=1:0.5的摩尔配比,再加入一定量的水,在800r/min的搅拌条件下,将硅溶胶、去离子水以及四乙基氢氧化铵混合,并于70℃的温度下水解10h,得到澄清的硅溶胶的水解溶液。然后在300r/min的搅拌条件下,按照矿化剂:SiO2=2.5:1的摩尔配比,在硅溶胶水解溶液中加入氟化钠和氟硅酸,搅拌均匀后得到H2O:OH-:SiO2=4:0.25:1的混合物。
将该混合物转移至密闭的耐压容器中,在700r/min的搅拌条件下,将晶化体系升温至100℃,并在自生压力下老化2天;随后继续在搅拌条件下将晶化体系升温至145℃,在自生压力下恒温9天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在140℃的温度下干燥5h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在800℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为3h,得到尺寸不大于490nm的全硅β分子筛。
实施例8
按SiO2:TEAOH=1:0.4的摩尔配比,再加入一定量的水,在100r/min的搅拌条件下,将硅胶、去离子水以及四乙基氢氧化铵混合,并于70℃的温度下水解10h,得到澄清的硅胶的水解溶液。在200r/min的搅拌条件下,按照矿化剂:SiO2=5:1的摩尔配比,在硅胶水解溶液中加入氯化钠和氢氟酸,搅拌均匀后得到OH-:SiO2=0.24:1的混合物。
将该混合物转移至密闭的耐压容器中,在100r/min的搅拌条件下,将晶化体系升温至80℃,并在自生压力下老化15天;随后继续在搅拌条件下将晶化体系升温至160℃,在自生压力下恒温30天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在180℃的温度下干燥0.5h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在700℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为1h,得到尺寸不大于490nm的全硅β分子筛。
对比例1
本对比例说明未按照本发明的技术方案,而是采用现有的中性含氟体系下(ChemCommun,1996,20:2365-2366.)全硅β分子筛的合成方法合成的全硅β分子筛的效果。
按SiO2:TEAOH:H2O=1:0.54:7.25的摩尔配比,在600r/min的搅拌条件下,将正硅酸乙酯、去离子水以及浓度为25.0重量%的四乙基氢氧化铵混合,并于70℃的温度下水解10h,得到澄清的正硅酸乙酯的水解溶液。在1000r/min的搅拌条件下,按照TEAOH:HF=1:1的摩尔配比,在正硅酸乙酯水解溶液中加入浓度为40.0重量%的氢氟酸,搅拌均匀后得到固态的硅胶。将硅胶研磨均匀,并转移至密闭的耐压容器中。
在100r/min的搅拌条件下,将晶化体系升温至135℃,在自生压力下恒温20天,得到晶化产物的混合物;将此混合物过滤,用水洗涤数遍后;在110℃的温度下干燥8h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在550℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为4h,得到全硅β分子筛,其X射线衍射(XRD)的晶相图如图6所示,扫描电子显微镜(SEM)的形貌结果如图7所示。
对比例2
本对比例说明未按照本发明的技术方案,而是采用现有的碱性条件下全硅β分子筛的合成方法(MicroporousMesoporousMater,2006,93:55-61.)合成的全硅β分子筛的效果。
按SiO2:TEAOH:H2O=1:0.4:9的摩尔配比,在400r/min的搅拌条件下,将白炭黑、去离子水以及四乙基氢氧化铵混合均匀,并于70℃的温度下水解10h,得到澄清的白炭黑水解溶液,随后按照TEA2SiF6:SiO2=0.07摩尔配比,加入TEA2SiF6并搅拌均匀。
在25℃老化24h后,将该混合溶液转移至密闭的耐压容器中,并在200r/min的搅拌条件下,将晶化体系升温至150℃,在自生压力下恒温13天,得到晶化产物的混合物;将此混合物过滤,用水洗涤至PH在6~8之间;在110℃的温度下干燥8h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在550℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为4h,得到全硅β分子筛,其X射线衍射(XRD)的晶相图如图8所示,扫描电子显微镜(SEM)的形貌结果如图9所示,29SiNMR的结果如图10所示。
对比例3
本对比例说明未按照本发明的技术方案,而是采用现有的酸性含氟体系下(EP2236461)全硅β分子筛的合成方法合成的全硅β分子筛的效果。
按SiO2:TEAOH:H2O=1:0.54:7.25的摩尔配比,在600r/min的搅拌条件下,将正硅酸乙酯、去离子水以及浓度为25.0重量%的四乙基氢氧化铵混合,并于70℃的温度下水解10h,得到澄清的正硅酸乙酯的水解溶液。在1000r/min的搅拌条件下,按照TEAOH:HF=1:1.5的摩尔配比,在正硅酸乙酯水解溶液中加入浓度为40.0重量%的氢氟酸,搅拌均匀后得到固态的硅胶。将硅胶研磨均匀,并转移至密闭的耐压容器中。
在100r/min的搅拌条件下,将晶化体系升温至135℃,在自生压力下恒温20天,得到晶化产物的混合物;将此混合物过滤,用水洗涤数遍后;在110℃的温度下干燥8h,将干燥的混合物研磨均匀后得到未焙烧的全硅β分子筛的原粉;最后,在550℃的温度下对全硅β分子筛的原粉进行焙烧处理,焙烧时间为4h,得到全硅β分子筛,其X射线衍射(XRD)的晶相图如图11所示,扫描电子显微镜(SEM)的形貌结果如图12所示。
实施例2-8的X射线衍射(XRD)晶相图与实施例1的类似(证明其为全硅β分子筛),故未一一列出;实施例1-8及对比例1-3中得到的beta分子筛通过SEM表征得到的长宽尺寸、厚度、形貌和29SiNMR表征得到的Q4/Q3值以及BET表征得到的比表面积SBET已系统列入表1中,具体结果如表1所示。
表1
从表1的数据可以看出,按照本发明的方法在碱性条件下采用分步晶化法进行水热晶化可以合成晶粒完好、均匀的纳米全硅beta分子筛,且得到的产物具有片状形貌,颗粒尺寸小,其长宽尺寸均不大于490纳米,厚度不大于150纳米,Q4/Q3不小于25说明骨架缺陷少、晶型完整,同时具有较大的比表面积,SBET均大于430m2/g。
以上结合附图详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
一种纳米全硅beta分子筛及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0