






专利摘要
本发明提供了一种N,O-双负离子配位的锆金属化合物及其制备方法和应用。所述的N,O-双负离子配位的锆金属化合物的制备方法:先将取代的水杨醛与二苯甲胺缩合,经硼氢化钠还原得到N,O-二齿配体,再用2倍摩尔量的正丁基锂去氢,与等摩尔量的氯化锆反应制得N,O-双负离子配位的锆金属化合物。本发明提供的N,O-双负离子配位的锆金属化合物能催化乙烯聚合制备超高分子量的聚乙烯,所得聚乙烯的重均分子量为(1.45~2.32)×106g/mol,分子量分布为3.32~36.6。
权利要求
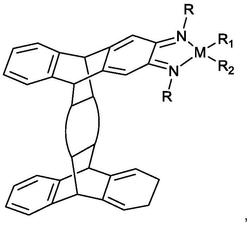
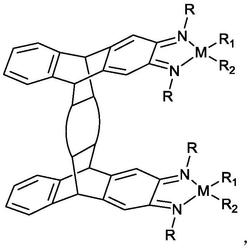
1.一种N,O-双负离子配位的锆金属化合物,其特征在于,具有如下结构式:
其中R1、R2可相同或不同,分别代表氢、烷基、烷氧基、芳基取代基团。
2.如权利要求1所述的N,O-双负离子配位的锆金属化合物的制备方法,其特征在于,包括如下步骤:
(1)N,O-二齿配体的制备:将取代的水杨醛溶于乙醇中,然后加入等摩尔量的二苯甲胺,室温搅拌12h,过滤得醛胺缩合产物;再将得到的醛胺缩合产物加到乙醇中,用冰水浴冷却至0℃,分批加入1.5倍摩尔量的硼氢化钠,控制加料速度使反应体系的温度保持在5-10℃,加完后室温搅拌12h;然后减压浓缩去除乙醇,将残留物倒入水中,用二氯甲烷提取,分出有机相,减压浓缩得N,O-二齿配体;
(2)氮气保护下,将步骤(1)得到的N,O-二齿配体溶于四氢呋喃中,用冰水浴冷却至0℃,然后滴加2倍摩尔量的正丁基锂溶液,控制滴加速度使反应体系的温度保持在0±2℃,加完后撤去冰水浴,室温搅拌反应6h;然后用丙酮浴降温至-78℃,分批加入等摩尔量的氯化锆,加完后使反应体系慢慢恢复至室温,搅拌反应12h;减压除去溶剂,残留物用二氯甲烷萃取,浓缩二氯甲烷萃取液,残留物用乙醚重结晶得四氢呋喃溶剂参与配位的N,O-双负离子配位的锆金属化合物。
3.如权利要求1所述的N,O-双负离子配位的锆金属化合物在催化乙烯聚合制备超高分子量聚乙烯中的应用。
说明书
技术领域
本发明涉及金属有机化合物技术领域,具体属于N,O-双负离子配位的锆金属化合物及其制备方法,以及该化合物在催化乙烯聚合反应中的应用。
背景技术
超高分子量聚乙烯是指分子量在100万以上的聚乙烯,具有许多普通聚乙烯和其他工程塑料不具备的优异特性,比如耐磨性极佳、冲击强度极高、耐低温、自润滑性好等优点,在某些领域已经取代了金属的地位,甚至其性能比金属制品更优良,因此,超高分子量聚乙烯被广泛应用于煤炭、建筑、军事、化工、体育、医疗、纺织、造纸、农业等领域。然而制备超高分子量聚乙烯的核心技术是烯烃聚合催化剂,用于烯烃聚合的催化剂有:传统Ziegler-Natta催化剂、茂金属催化剂、非茂金属催化剂等。虽然催化剂种类很多,但是已经实现工业化的催化剂很少,目前文献报道的制备超高分子量聚乙烯的催化剂大多以改进的Ziegler-Natta催化剂为主,存在聚合活性不高、聚合分子量不高、分子量分布较宽等问题,得到的超高分子量聚乙烯产品满足不了国民经济各个领域发展的需求。因此,研发新的烯烃聚合催化剂,改进烯烃聚合工艺,仍然是超高分子量聚乙烯研究领域的热点。
发明内容
本发明的目的是提供一种N,O-双负离子配位的锆金属化合物及其制备方法。
本发明的另一目的是提供一种N,O-双负离子配位的锆金属化合物的应用,即在催化乙烯聚合制备超高分子量聚乙烯中的应用。
本发明提供的一种N,O-双负离子配位的锆金属化合物,其结构式如下:
其中R1、R2可相同或不同,分别代表氢、烷基、烷氧基、芳基等取代基团。
本发明提供的一种N,O-双负离子配位的锆金属化合物的制备方法,是先将取代的水杨醛与二苯甲胺缩合,经硼氢化钠还原得到N,O-二齿配体,再用2倍摩尔量的正丁基锂去氢,与等摩尔量的氯化锆反应制得N,O-双负离子配位的锆金属化合物。其制备过程的反应式如下:
本发明的制备方法具体如下:
(1)N,O-二齿配体的制备:将取代的水杨醛溶于乙醇中,然后加入等摩尔量的二苯甲胺,室温搅拌12h,过滤得醛胺缩合产物。再将得到的醛胺缩合产物加到乙醇中,用冰水浴冷却至0℃,分批加入1.5倍摩尔量的硼氢化钠,控制加料速度使反应体系的温度保持在5-10℃,加完后室温搅拌12h。然后减压浓缩去除乙醇,将残留物倒入水中,用二氯甲烷提取,分出有机相,减压浓缩得N,O-二齿配体。
(2)氮气保护下,将步骤(1)得到的N,O-二齿配体溶于四氢呋喃中,用冰水浴冷却至0℃,然后滴加2倍摩尔量的正丁基锂溶液,控制滴加速度使反应体系的温度保持在0±2℃,加完后撤去冰水浴,室温搅拌反应6h。然后用丙酮浴降温至-78℃,分批加入等摩尔量的氯化锆,加完后使反应体系慢慢恢复至室温,搅拌反应12h。减压除去溶剂,残留物用二氯甲烷萃取,浓缩二氯甲烷萃取液,残留物用乙醚重结晶得四氢呋喃溶剂参与配位的N,O-双负离子配位的锆金属化合物。
本发明的N,O-双负离子配位的锆金属化合物可应用于催化乙烯聚合制备超高分子量的聚乙烯。催化乙烯聚合反应在高压反应釜中进行,聚合前反应釜需充分干燥,并通过抽真空、充乙烯气置换确保反应釜内的气体是纯净的乙烯气体。具体实验步骤为:在反应釜中,加入N,O-双负离子配位的锆金属化合物和甲苯,再加入助催化剂甲基铝氧烷(MAO),在10atm下搅拌反应。反应结束后,用盐酸酸化的乙醇溶液终止反应,过滤得超高分子量的聚乙烯。
本发明的优点与有益效果:
(1)本发明提供的N,O-双负离子配位的锆金属化合物,原料易得,制备方便。
(2)本发明提供的N,O-双负离子配位的锆金属化合物能催化乙烯聚合制备超高分子量的聚乙烯,所得聚乙烯的重均分子量为(1.45~2.32)×106g/mol。
附图说明
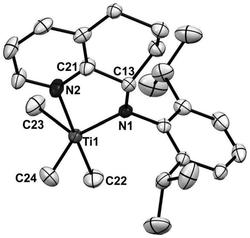
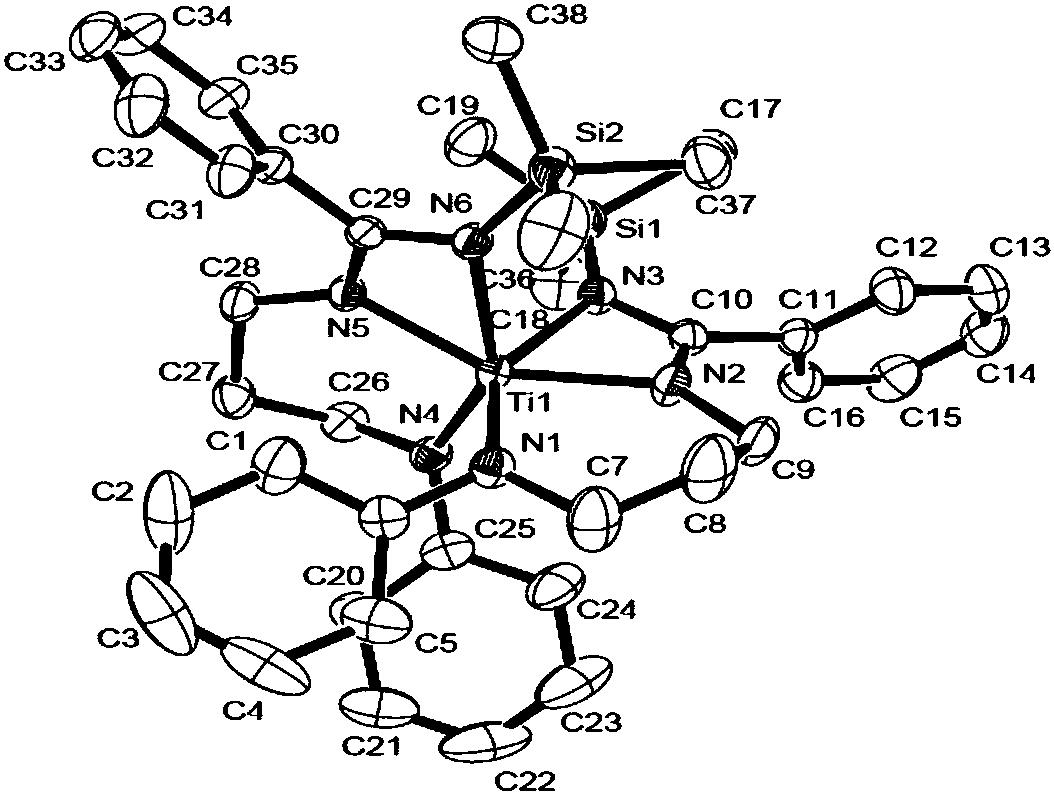
图1实施例1中所述的N,O-双负离子配位的锆金属化合物的晶体结构图。
具体实施方式
实施例1N,O-双负离子配位的锆金属化合物的制备
(1)2-(二苯甲胺基甲基)-4,6-二叔丁基苯酚配体的制备
将9.38g(40mmol)3,5-二叔丁基水杨醛溶于150mL乙醇中,搅拌下,加入7.33g(40mmol)二苯甲胺,加完后室温搅拌12h。反应体系中析出大量黄色固体,过滤,乙醇洗涤得醛胺缩合产物。将得到的醛胺缩合产物加到150mL乙醇中,用冰水浴冷却至0℃,然后分批加入2.27g(60mmol)硼氢化钠,控制加料速度使反应体系的温度保持在5-10℃,加完后室温搅拌12h。然后减压浓缩去除乙醇,将残留物加到50mL水中,用二氯甲烷提取,分出有机相,减压浓缩,得配体12.3g,产率76.6%。1HNMR(300MHz,DMSO-d6,ppm):δ11.94(br,1H,OH),7.49–7.39(m,8H,Ph-H),7.33(m,2H,Ph-H),7.17(s,1H,Ar-H),6.81(s,1H,Ar-H),4.97(d,J=9.30Hz,1H,CH),4.56(m,1H,NH),3.83(s,2H,CH2),1.44(s,9H,C(CH3)3),1.28(s,9H,C(CH3)3).13CNMR(75MHz,DMSO-d6,ppm):δ153.7(Ph),142.1(Ph),138.8(Ph),133.9(Ph),127.7(Ph),126.5(Ph),126.2(Ph),122.5(Ph),121.7(Ph),120.9(Ph),64.2(CH(Ph)2),50.1(CH2),33.7(C(Me)3),32.9(C(Me)3),30.8(Me),28.8(Me).
(2)氮气保护下,在反应瓶中,加入0.803g(2mmol)2-(二苯甲胺基甲基)-4,6-二叔丁基苯酚和20mL四氢呋喃,用冰水浴冷却至0℃,然后滴加1.9mL(4mmol)浓度为2.2M的正丁基锂溶液,控制滴加速度使反应体系的温度保持在0±2℃,加完后撤去冰水浴,室温搅拌反应6h。然后用丙酮浴降温至-78℃,分批加入0.466g(2mmol)氯化锆,加完后使反应体系慢慢恢复至室温,搅拌反应12h。减压除去溶剂,残留物用二氯甲烷萃取,浓缩二氯甲烷萃取液,残留物用乙醚重结晶得0.652g浅黄色四氢呋喃溶剂参与配位的N,O-双负离子配位的锆金属化合物,产率46%。
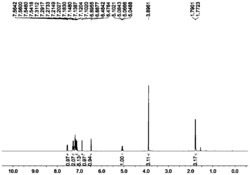
1HNMR(300MHz,CDCl3,ppm):δ7.22–6.99(m,12H,Ph-H,Ar-H),6.16(br,1H,CH),4.46(m,2H,CH2),4.06(m,8H,THF),1.81(m,8H,THF),1.47-1.40(m,9H,C(CH3)3),1.19-1.08(m,9H,C(CH3)3).
13CNMR(75MHz,CDCl3,ppm):δ152.7(Ph),144.8(Ph),142.2(Ph),135.9(Ph),130.2(Ph),129.9(Ph),129.3(Ph),128.3(Ph),126.5(Ph),121.3(Ph),68.9(THF),66.2(CH(Ph)2),48.3(CH2),36.2(C(Me)3),35.3(C(Me)3),32.4(Me),31.0(Me),26.6(THF).
晶体参数:化学式C36H49Cl2NO3Zr,分子量:705.88,单斜晶系,空间群P2(1)/c,晶胞参数 α=90.00°,β=112.323(3)°,γ=90.00°, Dx=1.284g/cm3,Z=4,R1=0.0369,wR2=0.0928,S=1.021。
X-射线晶体衍射结构图见图1。
以下实施例为N,O-双负离子配位的锆金属化合物催化乙烯聚合的应用实施例。催化乙烯聚合反应在500mL高压反应釜中进行,聚合前反应釜需充分干燥,并通过抽真空、充乙烯气置换三次,以确保反应釜内的气体是纯净的乙烯气体。
实施例2
在500mL反应釜中,加入3.5mg(5μM)实施例1制备的N,O-双负离子配位的锆金属化合物和100mL甲苯,再加入Al/Zr为1000:1的甲基铝氧烷(MAO)。在10atm下,30℃搅拌反应30min,用盐酸酸化的乙醇溶液终止反应,过滤,得到的聚合物用乙醇洗涤,干燥得聚合产物69mg,催化活性为2.76×104gPE/molZr·h。所得聚乙烯的重均分子量(Mw)为1.51×106g/mol,分子量分布为36.6。
实施例3
在500mL反应釜中,加入3.5mg(5μM)实施例1制备的N,O-双负离子配位的锆金属化合物和100mL甲苯,再加入Al/Zr为2000:1的MAO。在10atm下,30℃搅拌反应30min,用盐酸酸化的乙醇溶液终止反应,过滤,得到的聚合物用乙醇洗涤,干燥得聚合产物256mg,催化活性为1.02×105gPE/molZr·h。所得聚乙烯的重均分子量(Mw)为1.45×106g/mol,分子量分布为4.52。
实施例4
在500mL反应釜中,加入3.5mg(5μM)实施例1制备的N,O-双负离子配位的锆金属化合物和100mL甲苯,再加入Al/Zr为2500:1的MAO。在10atm下,30℃搅拌反应30min,用盐酸酸化的乙醇溶液终止反应,过滤,得到的聚合物用乙醇洗涤,干燥得聚合产物130mg,催化活性为5.20×104gPE/molZr·h。所得聚乙烯的重均分子量(Mw)为2.11×106g/mol,分子量分布为4.60。
实施例5
在500mL反应釜中,加入3.5mg(5μM)实施例1制备的N,O-双负离子配位的锆金属化合物和100mL甲苯,再加入Al/Zr为2000:1的MAO。在10atm下,50℃搅拌反应30min,用盐酸酸化的乙醇溶液终止反应,过滤,得到的聚合物用乙醇洗涤,干燥得聚合产物164mg,催化活性为6.56×104gPE/molZr·h。所得聚乙烯的重均分子量(Mw)为2.32×106g/mol,分子量分布为3.32。
实施例6
在500mL反应釜中,加入3.5mg(5μM)实施例1制备的N,O-双负离子配位的锆金属化合物和100mL甲苯,再加入Al/Zr为2000:1的MAO。在10atm下,70℃搅拌反应30min,用盐酸酸化的乙醇溶液终止反应,过滤,得到的聚合物用乙醇洗涤,干燥得聚合产物141mg,催化活性为5.64×104gPE/molZr·h。所得聚乙烯的重均分子量(Mw)为1.47×106g/mol,分子量分布为6.23。
实施例7
在500mL反应釜中,加入3.5mg(5μM)实施例1制备的N,O-双负离子配位的锆金属化合物和100mL甲苯,再加入Al/Zr为2000:1的MAO。在10atm下,30℃搅拌反应15min,用盐酸酸化的乙醇溶液终止反应,过滤,得到的聚合物用乙醇洗涤,干燥得聚合产物86mg,催化活性为6.88×104gPE/molZr·h。所得聚乙烯的重均分子量(Mw)为1.46×106g/mol,分子量分布为5.67。
实施例8
在500mL反应釜中,加入3.5mg(5μM)实施例1制备的N,O-双负离子配位的锆金属化合物和100mL甲苯,再加入Al/Zr为2000:1的MAO。在10atm下,30℃搅拌反应45min,用盐酸酸化的乙醇溶液终止反应,过滤,得到的聚合物用乙醇洗涤,干燥得聚合产物255mg,催化活性为6.80×104gPE/molZr·h。所得聚乙烯的重均分子量(Mw)为1.74×106g/mol,分子量分布为4.67。
N,O-双负离子配位的锆金属化合物及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0