









专利摘要
本发明涉及一种制备毫米级大粒度阿奇霉素的结晶方法;阿奇霉素结晶产品为白色晶体,产品形貌为一端有尖头的菱柱状,堆密度较好,达到0.58g/mL以上;流动性好,休止角可以小于36°;方法是将阿奇霉素粗品和丙酮溶剂加入到带搅拌的结晶器中加热溶解后,加入水达到过饱和后加入阿奇霉素晶种养晶,再滴加水让晶体生长,水滴加完后,过滤晶体,干燥后得到阿奇霉素晶体产品。本方法采用恒温溶析法,工艺流程简单,通过控制晶种和晶体的生长得到毫米级大粒度阿奇霉素产品,产品粒度大、没有破碎、纯度高、堆密度高、流动性好,容易实现工业化生产。
权利要求
1.一种毫米级大粒度阿奇霉素的结晶方法,其特征是:将一定配比的阿奇霉素粗品和丙酮溶剂加入到带搅拌的结晶器中加热溶解后,加入水达到过饱和后加入阿奇霉素晶种养晶,再滴加水让晶体生长,水滴加完后,过滤晶体,干燥后得到阿奇霉素晶体产品;
所述的阿奇霉素和丙酮溶剂的质量配比范围为1:2~5;所述的加入水达到过饱和,加入水的质量为阿奇霉素总质量的0.5~2倍;所述的滴加水让晶体生长,滴加水的质量为阿奇霉素总质量的2~6倍,滴加水的时间为5~10h;所述的晶种加入量为阿奇霉素总质量的0.5%~2%。
说明书
技术领域
本发明属于化学工程医药技术领域,具体涉及一种制备毫米级大粒度阿奇霉素的结晶方法
背景技术
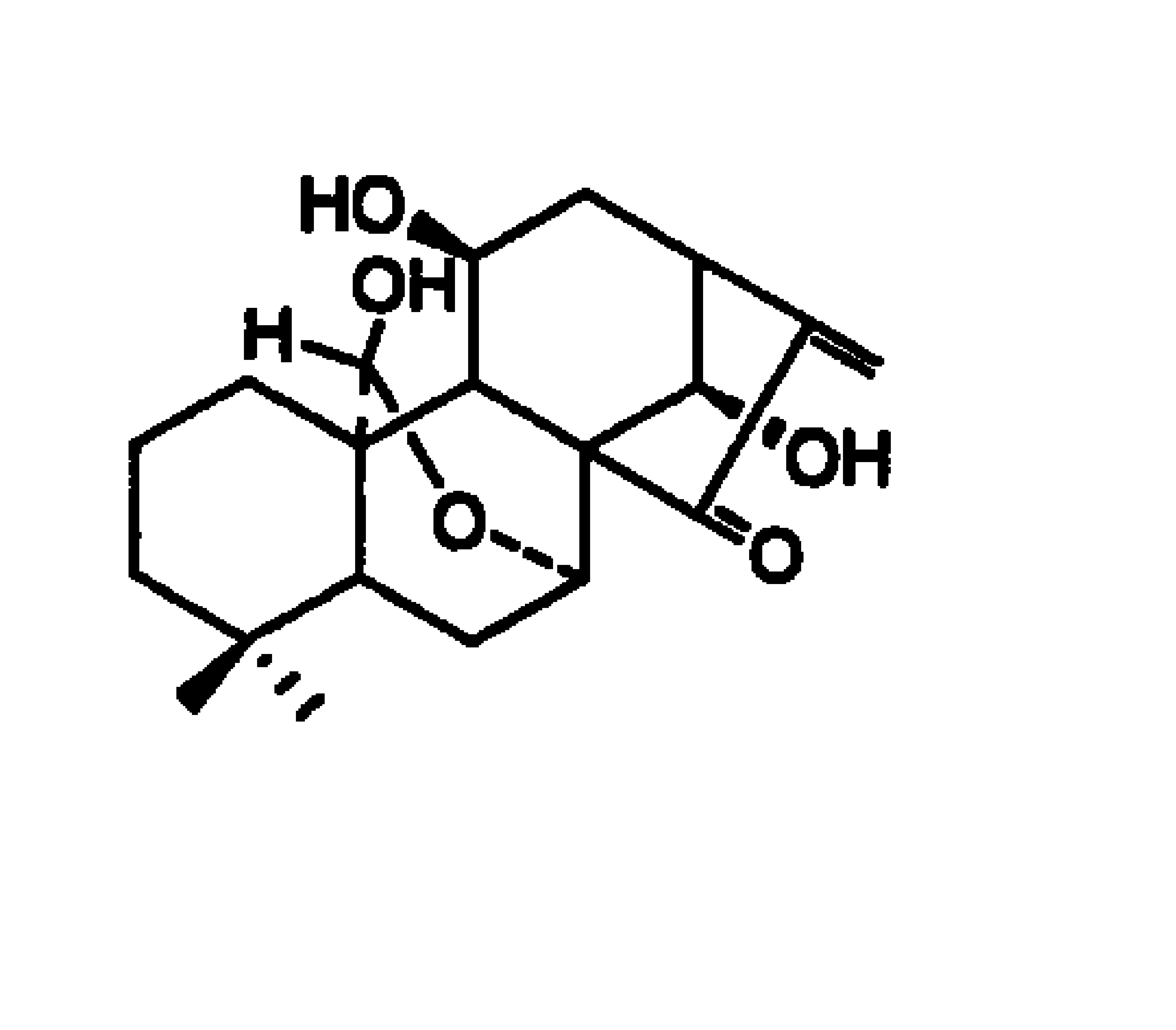
阿奇霉素(9-脱氧-9α-氮杂-9α-甲基-9α-红霉素A)是第三代大环内酯类抗生素。化学结构式为:
阿奇霉素通过阻碍细菌转肽过程,抑制细菌蛋白质合成而杀灭细菌。对革兰氏阳性菌、部分革兰氏阴性菌、衣原体、支原体等均有较强抗菌作用,对呼吸道感染疗效显著。阿奇霉素生物利用度高、体内抗菌活性强,组织渗透性强、疗效显著、安全性及受耐性好。美国专利US4517359和US4474768描述了阿奇霉素。
人体药动学表明,阿奇霉素生物半衰期非常长,具有很好的临床使用价值。阿奇霉素主要有片剂和注射剂,考虑用药方便性,片剂的适用性更好。而片剂对产品的粒度,堆密度和流动性的要求较高,阿奇霉素产品的这些粉体性能受精制和结晶方法影响很大。目前,国内外的报道主要关注在阿奇霉素的纯化精制方法,US0082527报道的用甲醇或丙酮和水的纯化精制方法;CN1304407C报道了吸附洗脱减压蒸馏和溶析的精制方法纯化阿奇霉素;CN104262429报道了通过萃取和酸碱中和反应纯化阿奇霉素;CN105001283B报道了通过乙醇和水结晶后再用丙酮和水重结晶的精制纯化阿奇霉素的方法;CN101418026B报道了一种晶型和粒度可控的结晶工艺,通过溶剂和酸碱反应速度,制备粒度范围在20~180微米的阿奇霉素产品。总结发现洗脱蒸发或者萃取酸碱中和反应的精制方法过程繁琐,难以实现工业化。而且这些方法都是针对阿奇霉素产品的纯度,并没有关注产品的粒度、形貌和聚结情况,目前市售的阿奇霉素原料基本上都是聚结体,产品堆密度小,流动性不好,堆密度在0.42g/mL~0.54g/mL,表征流动性的休止角一般大于42°。所以迫切需要我们开发和优化工艺得到堆密度较高,流动性较好的阿奇霉素产品。
目前工业上生产得到的阿奇霉素产品如图1所示,容易是聚结体,而且破碎很严重,粒度大小在30~200微米,粒度太小容易造成结块和聚结现象,聚结会导致溶剂保藏,纯度低;而且聚结和破碎会影响颗粒的流动性和堆密度,产品的流动性不好,堆密度也低。针对这些问题,需要开发一种工艺,使得产品粒度较大,不聚结,流动性和堆密度较高。
发明内容
为解决上述技术之不足,本发明的目的是提供一种制备毫米级大粒度阿奇霉素的结晶方法,所制备的阿奇霉素结晶产品为白色晶体,产品形貌为一端有尖头的菱柱状,晶体不聚结为单一分散的毫米级大粒度颗粒;产品纯度高可以达到99.8%;没有破碎现象;堆密度较好,可以达到0.58g/mL以上;流动性好,休止角可以小于36°;非常容易过滤和干燥;采用溶析结晶方法,结晶工艺简单,收率高,产品纯度高。
一种制备毫米级大粒度阿奇霉素的结晶方法,其特征在于:
将阿奇霉素粗品用丙酮加热溶解,加水达到过饱和,出晶前加入晶种后养晶,再继续滴加水溶析,晶体进一步生长成毫米级,经过滤干燥得到亮白色不聚结的毫米级阿奇霉素产品。
本发明的技术方案如下:
一种制备毫米级大粒度阿奇霉素的结晶方法,将阿奇霉素粗品和丙酮溶剂加入到带搅拌的结晶器中加热溶解后,加入水达到过饱和后加入阿奇霉素晶种养晶,再滴加水让晶体生长,水滴加完后,过滤晶体,干燥后得到阿奇霉素晶体产品。
所述的阿奇霉素和丙酮溶剂的质量比为1:2~5。
所述的加热溶解温度为30~50℃。
所述的加入水达到过饱和,加入水的质量为阿奇霉素总质量的0.5~2倍。
所述的晶种加入质量为阿奇霉素总质量的0.5%~2%;养晶10~40min。
所述的滴加水让晶体生长,滴加水的质量为阿奇霉素总质量的2~6倍;滴加水的时间为5~10h。
本发明所述的制备毫米级大粒度阿奇霉素的结晶方法制备的颗粒主粒度达到1.5毫米以上,产品纯度高,颗粒不聚结而且流动性非常好。在药物生产中,粒子的堆密度、粒子大小、流动性是很重要的粉体指标。堆密度增大、流动性提高有利于产品的储存、运输,所以能改善阿奇霉素粒子特性是结晶过程很重要的一项目标。通过本发明制备的阿奇霉素产品粒子是单分散不聚结的,产品主粒度可以达到1.5毫米以上;没有破碎现象;堆密度较好,可以达到0.58g/mL以上;流动性好,休止角可以小于38°。本发明中使用的阿奇霉素粗品纯度是95.6%,经过制备成毫米级大粒度产品后纯度高于99.5%,而且本发明的结晶方法是恒温溶析结晶,工艺简单,容易实现工业化生产。
附图说明
图1市售的阿奇霉素原料产品扫描电镜照片
图2实施例1所得产品的扫描电镜照片;
图3为实施例2所得产品的扫描电镜照片;
图4为实施例3所得产品的扫描电镜照片。
图5为实施例4所得产品的扫描电镜照片。
具体实施方法
以下将通过实施例形式的具体实施方式,具体步骤如下:在带搅拌的结晶器内加入阿奇霉素粗品与丙酮溶剂,1份质量的阿奇霉素粗品,加入丙酮的质量为2~5份,升高温度到30~50℃,确保阿奇霉素固体完全溶解,加入0.5~2份质量水后加入阿奇霉素总质量的0.5%~2%的阿奇霉素晶体作晶种养晶10~40min,继续滴加水2~6份质量的水,滴加时间控制为5~10h,搅拌速度控制在体系可以达到悬浮均匀即可,滴加结束后,过滤晶体,干燥得到阿奇霉素产品。对本发明的上述内容作进一步的详细说明。但不应该将此理解为本发明上述主题的范围仅限于以下实施例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
实施例1
将5g阿奇霉素和10g份丙酮加到带搅拌的结晶器中,加热到50℃保温,阿奇霉素全部溶解后,加入2.5g水,体系稳定后,加入阿奇霉素总重量的0.5%的阿奇霉素晶体当作晶种,恒温养晶30min,缓慢滴加10g水,整个滴加时间控制在大约5h,加完水后,过滤干燥得到产品。产品的SEM照片如附图2所示,从图中可以看出单个晶体产品的粒度大于1.4毫米,产品粒度分布均匀;堆密度0.58g/mL;休止角37.2°;通过液相检测产品纯度为99.7%。
实施例2
将10g阿奇霉素和50g丙酮加到带搅拌的结晶器中,温度控制在30℃保温,待阿奇霉素全部溶解后,加入20g水,体系稳定后,加入阿奇霉素总重量的2%的阿奇霉素晶体当作晶种,恒温养晶10min,缓慢滴加60g水,整个滴加时间控制在大约10h,加完水后,过滤干燥得到产品。产品的SEM照片如附图3所示,从图中可以看出单个晶体产品的粒度大于1.6毫米,产品粒度分布均匀;堆密度0.59g/mL;休止角36.5°;通过液相检测产品纯度为99.8%。
实施例3
将8g阿奇霉素和32g丙酮加到带搅拌的结晶器中,温度控制在50℃保温,待阿奇霉素全部溶解后,加入12g水,体系稳定后,加入阿奇霉素总重量的1%的阿奇霉素晶体当作晶种,恒温养晶40min,缓慢滴加40g水,整个滴加时间控制在大约8h,加完水后,过滤干燥得到产品。产品的SEM照片如附图4所示,从图中可以看出单个晶体产品的粒度大于1.5毫米,产品粒度分布均匀;堆密度0.60g/mL;休止角37.9°;通过液相检测产品纯度为99.8%。
实施例4
将6g阿奇霉素和18g丙酮加到带搅拌的结晶器中,温度控制在40℃保温,待阿奇霉素全部溶解后,加入6g水,体系稳定后,加入阿奇霉素总重量的1.5%的阿奇霉素晶体当作晶种,恒温养晶20min,缓慢滴加24g水,整个滴加时间控制在大约9h,加完水后,过滤干燥得到产品。单个晶体产品的粒度大于1.7毫米,产品粒度分布均匀;堆密度0.58g/mL;休止角38.0°;通过液相检测产品纯度为99.6%。
实施例5
将4g阿奇霉素和14g丙酮加到带搅拌的结晶器中,温度控制在35℃保温,待阿奇霉素全部溶解后,加入6.4g水,体系稳定后,加入阿奇霉素总重量的0.8%的阿奇霉素晶体当作晶种,恒温养晶25min,缓慢滴加22g水,整个滴加时间控制在大约7h,加完水后,过滤干燥得到产品。单个晶体产品的粒度大于1.4毫米,产品粒度分布均匀;堆密度0.57g/mL;休止角35.80°;通过液相检测产品纯度为99.8%。
实施例6
将10g阿奇霉素和25g丙酮加到带搅拌的结晶器中,温度控制在45℃保温,待阿奇霉素全部溶解后,加入8g水,体系稳定后,加入阿奇霉素总重量的1.2%的阿奇霉素晶体当作晶种,恒温养晶15min,缓慢滴加35g水,整个滴加时间控制在大约8h,加完水后,过滤干燥得到产品。单个晶体产品的粒度大于1.5毫米,产品粒度分布均匀;堆密度0.59g/mL;休止角36.50°;通过液相检测产品纯度为99.6%。
一种制备毫米级大粒度阿奇霉素的结晶方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0