







专利摘要
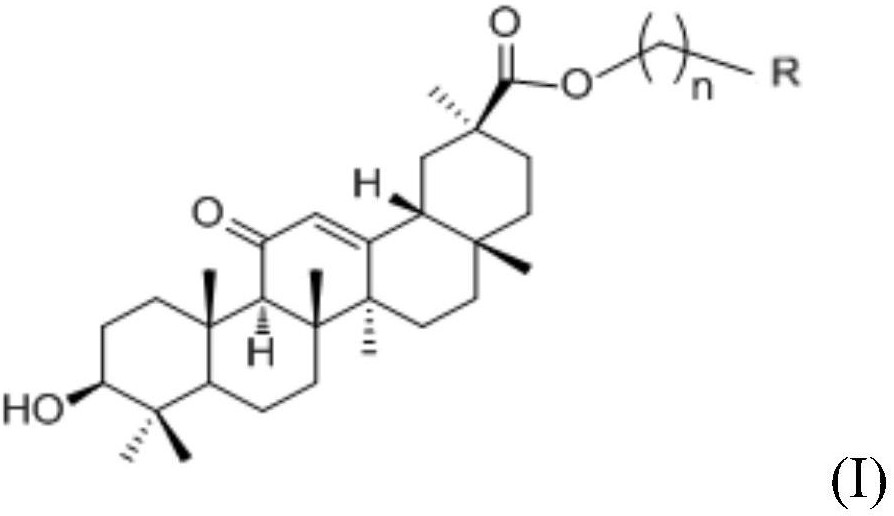
本发明公开了羊毛甾烷型三萜类化合物及其制备方法和用途,属于医药领域。本发明结构式为本发明化合物的分离制备方法是将毛果南烛叶的氯仿部位由正相硅胶柱划段,以CH2Cl2‑MeOH(50:1~0:1)进行洗脱,将得到的部分再进行正相CH2Cl2‑MeOH(40:1~0:1)、反相硅胶以MeOH‑H2O(20:1,40:1,60:1,80:1)洗脱划段,将得到的化合物再经凝胶、HPLC进行分离纯化。本发明化合物可用于制备抗肿瘤药物或作为抗肿瘤药物开发的先导化合物。
权利要求
1.羊毛甾烷型三萜类化合物,其特征在于所述的羊毛甾烷型三萜类化合物为
结构式Ⅰ中R
2.如权利要求1所述的羊毛甾烷型三萜类化合物的制备方法,其特征在于所述的制备方法是按下述步骤进行的:
步骤一、将毛果南烛叶干燥后粉碎,用95%(体积)乙醇提取3次,减压浓缩得到总浸膏;
步骤二、总浸膏加适量蒸馏水混悬,依次用石油醚、氯仿、乙酸乙酯和正丁醇萃取;
步骤三、步骤二萃取物的氯仿部位用硅胶按一定质量比(1:1)拌样,用CH
步骤四、将步骤三体积比为15:1的CH
按步骤三将体积比为12:1的CH
同时再收集其中20:1部分再经中压ODS柱子,以MeOH-H
3.根据权利要求2所述羊毛甾烷型三萜类化合物的制备方法,其特征在于步骤一中用95%乙醇提取的时间间隔为24h,温度为45℃,固液比为1:3。
4.根据权利要求2所述羊毛甾烷型三萜类化合物的制备方法,其特征在于将步骤三中将步骤二得到的氯仿提取物用100~200目硅胶按照质量1:1拌样,依次以CH
5.如权利要求1所述羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗肿瘤药物中的应用。
6.根据权利要求5所述羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备治疗白血病药物中的应用。
7.根据权利要求5所述羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗乳腺癌药物中的应用。
8.根据权利要求5所述羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗肝癌药物中的应用。
9.根据权利要求5所述羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗结肠癌药物中的应用。
10.根据权利要求5所述羊毛甾烷型三萜类化合物的用途,其特征在于所述羊毛甾烷型三萜类化合物在制备抗肺癌药物中的应用。
说明书
技术领域:
本发明属于医药技术领域;具体涉及羊毛甾烷型三萜类化合物及其制备方法和用途。
背景技术:
恶性肿瘤(malignant tumor),即癌症,是目前危害人类健康的主要疾病之一。近年来,我国肿瘤死亡率和发病率呈上升趋势,且发病患者越来越年轻,肿瘤死亡人数甚至占了全世界肿瘤死亡总数的24%。药物治疗在恶性肿瘤的三大疗法中占有重要的地位,目前,临床使用的抗肿瘤药物中绝大多数都是以天然抗肿瘤活性成分为先导化合物,经结构修饰或化学合成而来。
羊毛甾烷型三萜多分布于海参属和灵芝属中,在植物中极为罕见,仅在百合科,风信子科,马鞭草科,豆科和杜鹃花科植物中被发现过,杜鹃花科植物分布广泛,资源丰富,以往植物学家对其化学成分的研究主要集中在结构多样的二萜类化合物上,对三萜类化合物的研究并不十分全面。
发明内容:
本发明的目的是提供一种羊毛甾烷型三萜类化合物及其制备方法和用途。本发明从植物毛果南烛叶中分离提取的多种羊毛甾烷型三萜类化合物,提供了羊毛甾烷型三萜类化合物的制备方法。以顺铂(Cis-platinum MW 300)为阳性对照,本发明所述的三萜类化合物具有一定的抑制肿瘤细胞增殖活性,包括HL-60(人早幼粒白血病细胞)、MCF-7(乳腺癌细胞)、SMMC-7221(人肝癌细胞)、A-549(人肺腺癌细胞)、SW480(人结肠癌细胞)和BEAS-2B(肺上皮细胞)。这些化合物可用于制备抗肿瘤药物或作为抗肿瘤药物开发的先导化合物。
本发明的化合物结构式Ⅰ:
,其中化合物1-4中
本发明的化合物结构式Ⅱ:
,其中化合物5-7中 (R)表示C-24的绝对构型为順式。
本发明中羊毛甾烷型三萜类化合物的制备方法是按下述步骤进行的:
步骤一、将毛果南烛叶干燥后粉碎,用95%(体积)乙醇提取3次,减压浓缩得到总浸膏;
步骤二、总浸膏加适量蒸馏水混悬,依次用石油醚、氯仿、乙酸乙酯和正丁醇萃取;
步骤三、步骤二萃取物的氯仿部位用硅胶按一定质量比(1:1)拌样,用CH2Cl2-MeOH进行梯度(50:1→30:1→20:1→15:1→12:1→8:1→5:1→3:1→0:1)洗脱;
步骤四、将步骤三体积比为15:1的CH2Cl2-MeOH洗脱部位过正向硅胶柱划段,再用CH2Cl2-MeOH进行梯度(40:1→30:1→20:1→15:1→10:1→5:1→0:1)洗脱,取体积比为(15~0):1的CH2Cl2-MeOH洗脱部位,再经中压ODS(填料为YMC GEL ODS-AS 50μm,Japan)柱子,依次用20%(体积)MeOH-H2O、40%MeOH-H2O、60%MeOH-H2O、80%MeOH-H2O洗脱,将80%甲醇-水洗脱部位经Sephadex LH-20柱子用纯MeOH洗脱,每20mL作为一个单位接收,经TLC检测,合并向同样点接受液浓缩至干,再经半制备HPLC(戴安P680高效液相色谱仪,UVD170紫外检测器,RP C-18柱子(250×10nm,Welch))进一步分离,以流动相MeCN-H2O(80:20,v/v,流速:1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到3ɑ-O-ɑ-L-阿拉伯糖-24(S),25,30-三醇-羊毛甾-8-烯(tR=33.3min)、3α-O-β-D-葡萄糖-24(R),25,30-三醇-羊毛甾-8-烯(tR=20.6min)和3ɑ-O-β-D-葡萄糖-24(S),25,30-三醇-羊毛甾-8-烯tR=22.8min);
按步骤三将体积比为12:1的CH2Cl2-MeOH洗脱部位,过聚酰胺柱子(除掉黄酮类物质)用蒸馏水洗脱,合并所得水部位,再过正向硅胶柱划段,以CH2Cl2-MeOH(30:1→20:1→15:1→10:1→5:1→0:1)进行洗脱,收集其中15:1部分再经中压ODS柱子,以MeOH-H2O(20:1,40:1,60:1,80:1)洗脱,其中80%MeOH(20.4g)洗脱产品再经凝胶Sephadex LH-20分离,得到粗品每20mL作为一个单位接收,经TLC检测,合并相同样点接受液浓缩至干,然后经HPLC纯化,以流动相MeCN-H2O-CH3COOH(50:50:0.2,v/v/v,流速1.5mL/min)洗脱,检测波长设定为210nm,每次进样量为80μL,得到3α-O-β-D-葡萄糖-24(R),25-二醇-羊毛甾-8-烯-30-酸(tR=38.1min)。
同时再收集其中20:1部分再经中压ODS柱子,以MeOH-H2O(20:1,40:1,60:1,80:1)洗脱,其中80%MeOH洗脱产品再经凝胶Sephadex LH-20分离,得到粗品每20mL作为一个单位接收,经TLC检测,合并相同样点接受液浓缩至干,经HPLC纯化,以流动相MeCN-H2O-CH3COOH(70:30:0.1,v/v/v,流速1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到3-ɑ-O-ɑ-L-阿拉伯糖-羊毛甾-8-烯-24(S),25-二羟基-30-酸(tR=26.6min),24-ɑ-O-ɑ-L-阿拉伯糖-羊毛甾-8-烯-3ɑ,25-二羟基-30-酸(tR=25.5min),3-ɑ-O-ɑ-L-阿拉伯糖-羊毛甾-8-烯-24(R),25-二羟基-30-酸(tR=24.5min)。
上述羊毛甾烷型三萜类化合物在制备抗肿瘤药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备治疗白血病药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备抗乳腺癌药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备抗肝癌药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备抗结肠癌药物中的应用。
进一步限定,上述羊毛甾烷型三萜类化合物在制备抗肺癌药物中的应用。
本发明的羊毛甾烷型三萜具有6/6/6/5四环结构,通常含有8个甲基取代(C-4二甲基取代,C-10,C-13,C-14,C-20和C-25二甲基取代),天然植物中此类化合物结构稳定,新化合物较少见,本发明中的这类羊毛甾烷型三萜的C-24连羟基或糖,同时C-14位被氧化为醇羟基或羧基,极为罕见。
本发明首次从杜鹃花科植物L.ovalifolia var.hebecarpa.中发现羊毛甾烷型三萜类化合物并提供了这类新化合物的提取分离技术、结构鉴定方法和抗肿瘤细胞增殖方面的用途。
附图说明:
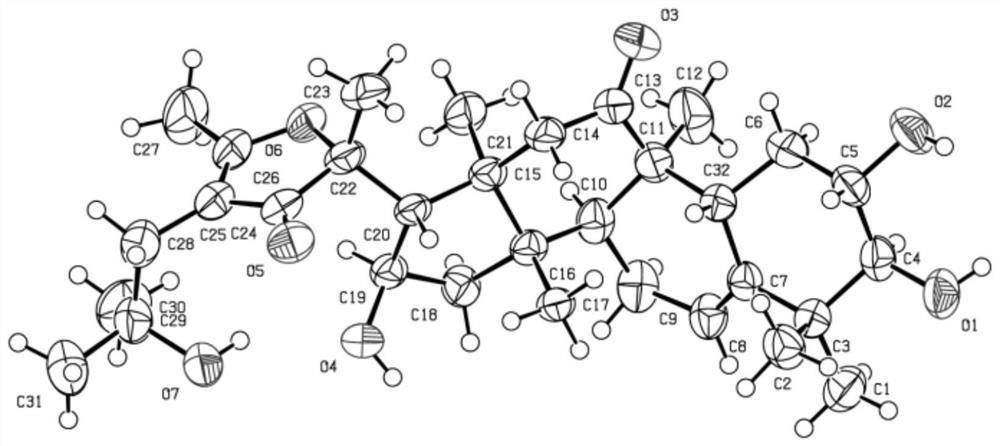
化合物1~4结构相似,仅在C-3,C-14和C-24上取代基存在差异,下面提供化合物1的的
图1是化合物1的
具体实施方式
实施例1:本实施例中羊毛甾烷型三萜类化合物制备方法是按下述步骤进行的:
步骤一、干燥毛果南烛叶50kg经粉碎,用95%乙醇(24h/次,220L,45C)提取3次,减压浓缩得到总浸膏9.15kg。
步骤二、将总浸膏加9.15kg蒸馏水混悬,依次用石油醚、氯仿、乙酸乙酯和正丁醇萃取,氯仿部位得到萃取物5.09kg。
步骤三、步骤二萃取物的氯仿部位用100~200目硅胶按照质量1:1拌样,以CH2Cl2-MeOH进行梯度(50:1→30:1→20:1→15:1→12:1→8:1→5:1→3:1→0:1)洗脱,得到9个部分。
步骤四、化合物1,2,5的制备:取步骤三体积比为15:1的CH2Cl2-MeOH洗脱部位,(129g)过聚酰胺柱子(除掉黄酮类物质)合并水部位35g,过正向硅胶柱划段,以CH2Cl2-MeOH(40:1→30:1→20:1→15:1→10:1→5:1→0:1)进行梯度洗脱,再经ODS(填料为YMC GELODS-AS 50μm,Japan)柱子,依次用20%(体积)MeOH-H2O、40%MeOH-H2O、60%MeOH-H2O、80%MeOH-H2O洗脱,其中将80%甲醇-水洗脱部位经Sephadex LH-20柱子用纯MeOH洗脱,每20mL作为一个单位接收,经TLC检测,合并相同样点接受液浓缩至干,粗品(142mg)再经半制备HPLC(戴安P680高效液相色谱仪,UVD170紫外检测器,RP C-18柱子(250×10nm,Welch))进一步分离,以流动相MeCN-H2O(80:20,v/v,流速:1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到3ɑ-O-ɑ-L-阿拉伯糖-24(S),25,30-三醇-羊毛甾-8-烯(3Mg,tR=33.3min)、3α-O-β-D-葡萄糖-24(R),25,30-三醇-羊毛甾-8-烯(7mg tR=20.6min)和3ɑ-O-β-D-葡萄糖-24(S),25,30-三醇-羊毛甾-8-烯(20mg,tR=22.8min);
化合物6的制备:将步骤三体积比为12:1的CH2Cl2-MeOH洗脱部位,过聚酰胺柱子(除掉黄酮类物质)用蒸馏水洗脱,合并所得水部位,再过正向硅胶柱划段,以CH2Cl2-MeOH(30:1→20:1→15:1→10:1→5:1→0:1)进行洗脱,收集其中15:1部分再经中压ODS柱子,以MeOH-H2O(20:1,40:1,60:1,80:1)洗脱,其中80%MeOH(20.4g)洗脱产品再经凝胶SephadexLH-20分离,得到粗品每20mL作为一个单位接收,经TLC检测,合并相同样点接受液浓缩至干,得到粗品(2.2g)取其中200mg经HPLC纯化,以流动相MeCN-H2O-CH3COOH(50:50:0.2,v/v/v,流速1.5mL/min)洗脱,检测波长设定为210nm,每次进样量为80μL,得到3α-O-β-D-葡萄糖-24(R),25-二醇-羊毛甾-8-烯-30-酸(7.6mg,tR=38.1min);
化合物3,4,7的制备:同时再收集其中20:1部分再经中压ODS柱子,以MeOH-H2O(20:1,40:1,60:1,80:1)洗脱,其中80%MeOH洗脱产品再经凝胶SephadexLH-20分离,得到粗品每20mL作为一个单位接收,经TLC检测,合并相同样点接受液浓缩至干,经HPLC纯化,以流动相MeCN-H2O-CH3COOH(70:30:0.1,v/v/v,流速1.5mL/min),检测波长设定为210nm,每次进样量为80μL,得到3-ɑ-O-ɑ-L-阿拉伯糖-羊毛甾-8-烯-24(S),25-二羟基-30-酸(12mg,tR=26.6min),24-ɑ-O-ɑ-L-阿拉伯糖-羊毛甾-8-烯-3ɑ,25-二羟基-30-酸(7mg,tR=25.5min),3-ɑ-O-ɑ-L-阿拉伯糖-羊毛甾-8-烯-24(R),25-二羟基-30-酸(120mg,tR=24.5min)。
通过NMR、化学反应方法确定化合物1,2,3,4,5,6,7的绝对构型。
化合物1:白色粉末;-85(c 0.4,MeOH);
化合物2:白色粉末;-22(c 0.1,MeOH);
化合物3:白色粉末;-12(c 0.4,MeOH);
化合物4:白色粉末;-20(c 1.1,MeOH);
化合物5:白色粉末;-15(c 0.3,MeOH);
化合物6:白色粉末;-7(c 0.4,MeOH);
化合物7:白色粉末;-65(c 1.5,MeOH);
化合物1,2,3,4,5,6,7的
Table 1.
Table 2.
化合物的糖苷键的酸水解
为了确定化合物1中阿拉伯糖的绝对构型,将其进行酸水解,水层得阿拉伯粗多糖。向化合物1酸水解得到的阿拉伯糖与D-(1mg)、L-阿拉伯糖(1mg)标准品中分别加入L-半胱氨酸甲酯盐酸盐和无水吡啶于65℃下反应2h,随后再加入N-三甲基硅咪唑继续反应1.5h使其分别硅烷化,减压蒸干,加入蒸馏水和正己烷萃取,有机层分别得到化合物1中阿拉伯糖的硅烷化衍生物和标准L-、D-阿拉伯糖的硅烷化衍生物。利用GC检测,化合物1中阿拉伯糖硅烷衍生物、标准L-、D-阿拉伯糖的硅烷化衍生物的保留时间分别为8.83、8.82和9.13min,确定化合物1中的糖为L-阿拉伯糖。
利用GC检测,化合物2,3,5中阿拉伯糖硅烷衍生物的保留时间分别为8.88,8.90,and 8.84min,确定化合物2,3,5中的糖为L-阿拉伯糖。
化合物4的酸水解:将化合物4(6mg)溶于2mL甲醇中,加入2mol/L的HCl水溶液2mL,70℃下水解96h,反应液减压蒸干至中性,加入1mL蒸馏水混匀,再用1mL乙酸乙酯萃取3次。水层减压浓缩蒸干得到粗单糖(0.5mg)。
单糖的衍生化:向水解得到的单糖(0.5mg)、D-(1mg)和L-葡萄糖标准品(1mg)中分别加入2mg的L-半胱氨酸甲酯盐酸盐,0.2mL的无水吡啶,65℃下反应2h,随后在混合液中加入0.2mLN-三甲基硅咪唑65℃下继续反应1.5h,减压蒸干,加入1mL蒸馏水,0.2mL正己烷萃取,有机层用GC检测。
GC检测条件:WM-1毛细管柱(30m*0.25mm*0.5μm,Welch);柱温:220℃保留5min,每分钟5℃升温到270℃,进样口温度:250℃,载气:N2,1.0mL/min,分流比:10:1;FID检测器,检测温度:250℃。
化合物4中糖衍生物经GC分析,保留时间为13.30min,D-和L-葡萄糖标准品的衍生化产物的保留时间分别为13.35和14.85min,因此,化合物4的糖为D-葡萄糖。
利用GC检测,化合物6,7中葡萄糖硅烷衍生物的保留时间分别为13.34,和13.35min,确定化合物6,7中的糖为D-葡萄糖。
实施例2化合物3,4,7对HL-60(人早幼粒白血病细胞)、MCF-7(乳腺癌细胞)、SMMC-7221(人肝癌细胞)、A-549(人肺腺癌细胞)、SW480(人结肠癌细胞)和BEAS-2B(肺上皮细胞)五种肿瘤细胞增殖的抑制活性,这三种化合物对正常细胞均无毒副作用(IC50>40μM)。且其中化合物3和4抑制A-549肿瘤细胞增殖活性优于阳性对照药物顺铂,化合物3和7抑制MCF-7肿瘤细胞增殖活性也优于阳性对照药物顺铂,具体数据见表3
Table 3.Anti-proliferative Activities of Compounds 1-7against FiveCancer Cell Linesand One Normal cell line(IC50inμM)
实验结论:羊毛甾烷型三萜单糖苷,C-14位存在-CH2OH取代时具有不同抑制肿瘤细胞增殖活性,C-3位的糖部分取代基团差异和C-24位的构型对化合物抗肿瘤活性的强弱程度及选择性有较大影响。
利用MTS法,研究这7个新化合物对HL-60(人早幼粒白血病细胞)、MCF-7(乳腺癌细胞)、SMMC-7721(人肝癌细胞)、A-549(人肺腺癌细胞)、SW480(人结肠癌细胞)和BEAS-2B(肺上皮细胞)五种肿瘤细胞增殖抑制活性,本发明发现化合物3有抑制HL-60,A-549,SMMC-7721,MCF-7,SW480五种肿瘤细胞增殖的活性,化合物4有抑制HL-60,A-549,SMMC-7721三种肿瘤细胞增殖的活性,化合物7有抑制HL-60,SMMC-7721,MCF-7三种肿瘤细胞增殖的活性。
本发明对HL-60,A-549,SMMC-7721,MCF-7,SW480五种肿瘤细胞增殖的抑制活性。化合物1,2,3,4,5,6,7均对正常细胞BEAS-2B无毒副作用,表示其具有显著的抗肿瘤活性。
羊毛甾烷型三萜类化合物及其制备方法和用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0