









专利摘要
本发明公开了一种制备雷公藤甲素的方法。存在于雷公藤中的雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2‑表雷公藤乙素结构非常接近,其中雷公藤乙素和2‑表雷公藤乙素为一对手性异构体,分离难度大。现有技术在分离这四种化合物时,无不依赖于反复硅胶柱层析,然而反复硅胶柱层析耗费溶剂、重现性差,只适合于实验室少量分离,难以在工业生产中推广。目前工业生产中,唯一在使用的柱色谱只有大孔树脂。本发明提供的方法不依赖于反复硅胶柱层析即可分离制备雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2‑表雷公藤乙素。本发明提供的HPLC方法基于常规的C18色谱柱即可有效分离手性异构体雷公藤乙素和2‑表雷公藤乙素,成本低、重复性高。
权利要求
1.一种制备雷公藤甲素的方法,其特征在于,包括如下步骤:
步骤S1,提取:将卫矛科植物雷公藤的干燥根粉碎,乙醇溶液冷浸提取,回收提取液中的乙醇并用水稀释,过滤,收集滤液作为大孔树脂上样液;
步骤S2,XDA-1B大孔吸附树脂富集:以XDA-1B大孔吸附树脂为分离介质并装柱,树脂径高比为1:10,拌样树脂占树脂总量的1/10,湿法装柱,拌树脂上样;先用12BV的35%乙醇以12BV/h的流速清洗,后用8BV的75%乙醇(含万分之一三乙胺,体积百分含量)以12BV/h的流速洗脱并收集5-8BV洗脱液;将收集的洗脱液浓缩至干即得粗品;
步骤S3,碱开环酸闭环精制:将步骤S2所得粗品使用pH值为9.5的碱水溶解,过滤去除不溶物,滤液再用酸调节pH值至6.5,低温析晶,过滤收集析出物,洗涤、干燥,用65%乙醇(含万分之一三乙胺,体积百分含量)溶解作为上样液;
步骤S4,DM130大孔吸附树脂分离:以DM130大孔吸附树脂为分离介质并装柱,树脂径高比为1:20,拌样树脂占树脂总量的1/20,湿法装柱,拌树脂上样;用12BV的65%乙醇(含万分之一三乙胺,体积百分含量)以4BV/h的流速洗脱并收集11-12BV洗脱液,11-12BV洗脱液浓缩干燥即得雷公藤甲素。
2.根据权利要求1所述的方法,其特征在于:步骤S1提取的固液比为1:5,1kg干燥根对应用5L的乙醇溶液冷浸提取。
3.根据权利要求1或2所述的方法,其特征在于:步骤S1用95%乙醇冷浸提取。
4.根据权利要求1所述的方法,其特征在于:步骤S1回收提取液中的乙醇后用水稀释至0.5g生药/mL,过滤,收集滤液作为大孔树脂上样液。
5.根据权利要求1所述的方法,其特征在于:步骤S2拌样树脂质量为上样液对应生药质量的1/2。
6.根据权利要求1所述的方法,其特征在于:步骤S3碱水为氨水溶液。
7.根据权利要求1所述的方法,其特征在于:步骤S3酸为盐酸。
8.根据权利要求1所述的方法,其特征在于:步骤S3低温析晶的温度为4℃。
9.根据权利要求1所述的方法,其特征在于:步骤S3用65%乙醇(含万分之一三乙胺,体积百分含量)溶解成0.5g/mL溶液作为上样液。
10.根据权利要求1所述的方法,其特征在于:步骤S4拌样树脂质量为上样液对应析出物质量的1/2。
说明书
技术领域
本发明属于化学领域,涉及已知化合物的制备方法和分离分析方法,具体涉及一种制备雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素的方法以及雷公藤乙素和2-表雷公藤乙素的HPLC分离分析方法。
背景技术
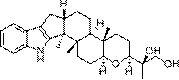
研究发现,存在于雷公藤中的四种结构非常类似的化学成分具有优异的抗肿瘤活性,分别为:雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素。结构式如下:
从化学结构可以发现,这四个化合物结构非常接近,其中雷公藤乙素和2-表雷公藤乙素为一对手性异构体。这导致这四个化合物的分离难度非常大。现有技术在分离这四种化合物时,无不依赖于反复硅胶柱层析,然而反复硅胶柱层析耗费溶剂、重现性差,只适合于实验室少量分离,难以在工业生产中推广。目前工业生产中,唯一在使用的柱色谱只有大孔树脂。
因此,一种基于大孔树脂的分离方法有助于实现上述四种化合物的工业化生产制备。
发明内容
本发明的目的在于克服现有技术的不足,提供一种制备雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素的方法及雷公藤乙素和2-表雷公藤乙素的HPLC分离分析方法。
本发明的上述目的是通过下面的技术方案得以实现的:
一种制备雷公藤内酯酮的方法,包括如下步骤:
步骤S1,提取:将卫矛科植物雷公藤的干燥根粉碎,乙醇溶液冷浸提取,回收提取液中的乙醇并用水稀释,过滤,收集滤液作为大孔树脂上样液;
步骤S2,XDA-1B大孔吸附树脂富集:以XDA-1B大孔吸附树脂为分离介质并装柱,树脂径高比为1:10,拌样树脂占树脂总量的1/10,湿法装柱,拌树脂上样;先用12BV的35%乙醇以12BV/h的流速清洗,后用8BV的75%乙醇(含万分之一三乙胺,体积百分含量)以12BV/h的流速洗脱并收集5-8BV洗脱液;将收集的洗脱液浓缩至干即得粗品;
步骤S3,碱开环酸闭环精制:将步骤S2所得粗品使用pH值为9.5的碱水溶解,过滤去除不溶物,滤液再用酸调节pH值至6.5,低温析晶,过滤收集析出物,洗涤、干燥,用65%乙醇(含万分之一三乙胺,体积百分含量)溶解作为上样液;
步骤S4,DM130大孔吸附树脂分离:以DM130大孔吸附树脂为分离介质并装柱,树脂径高比为1:20,拌样树脂占树脂总量的1/20,湿法装柱,拌树脂上样;用9BV的65%乙醇(含万分之一三乙胺,体积百分含量)以4BV/h的流速洗脱并收集8-9BV洗脱液,8-9BV洗脱液浓缩干燥即得雷公藤内酯酮。
优选地,步骤S1提取的固液比为1:5,1kg干燥根对应用5L的乙醇溶液冷浸提取。
优选地,步骤S1用95%乙醇冷浸提取。
优选地,步骤S1回收提取液中的乙醇后用水稀释至0.5g生药/mL,过滤,收集滤液作为大孔树脂上样液。
优选地,步骤S2拌样树脂质量为上样液对应生药质量的1/2。
优选地,步骤S3碱水为氨水溶液。
优选地,步骤S3酸为盐酸。
优选地,步骤S3低温析晶的温度为4℃。
优选地,步骤S3用65%乙醇(含万分之一三乙胺,体积百分含量)溶解成0.5g/mL溶液作为上样液。
优选地,步骤S4拌样树脂质量为上样液对应析出物质量的1/2。
一种制备雷公藤甲素的方法,包括如下步骤:
步骤S1,提取:将卫矛科植物雷公藤的干燥根粉碎,乙醇溶液冷浸提取,回收提取液中的乙醇并用水稀释,过滤,收集滤液作为大孔树脂上样液;
步骤S2,XDA-1B大孔吸附树脂富集:以XDA-1B大孔吸附树脂为分离介质并装柱,树脂径高比为1:10,拌样树脂占树脂总量的1/10,湿法装柱,拌树脂上样;先用12BV的35%乙醇以12BV/h的流速清洗,后用8BV的75%乙醇(含万分之一三乙胺,体积百分含量)以12BV/h的流速洗脱并收集5-8BV洗脱液;将收集的洗脱液浓缩至干即得粗品;
步骤S3,碱开环酸闭环精制:将步骤S2所得粗品使用pH值为9.5的碱水溶解,过滤去除不溶物,滤液再用酸调节pH值至6.5,低温析晶,过滤收集析出物,洗涤、干燥,用65%乙醇(含万分之一三乙胺,体积百分含量)溶解作为上样液;
步骤S4,DM130大孔吸附树脂分离:以DM130大孔吸附树脂为分离介质并装柱,树脂径高比为1:20,拌样树脂占树脂总量的1/20,湿法装柱,拌树脂上样;用12BV的65%乙醇(含万分之一三乙胺,体积百分含量)以4BV/h的流速洗脱并收集11-12BV洗脱液,11-12BV洗脱液浓缩干燥即得雷公藤甲素。
优选地,步骤S1提取的固液比为1:5,1kg干燥根对应用5L的乙醇溶液冷浸提取。
优选地,步骤S1用95%乙醇冷浸提取。
优选地,步骤S1回收提取液中的乙醇后用水稀释至0.5g生药/mL,过滤,收集滤液作为大孔树脂上样液。
优选地,步骤S2拌样树脂质量为上样液对应生药质量的1/2。
优选地,步骤S3碱水为氨水溶液。
优选地,步骤S3酸为盐酸。
优选地,步骤S3低温析晶的温度为4℃。
优选地,步骤S3用65%乙醇(含万分之一三乙胺,体积百分含量)溶解成0.5g/mL溶液作为上样液。
优选地,步骤S4拌样树脂质量为上样液对应析出物质量的1/2。
一种制备雷公藤乙素和2-表雷公藤乙素的方法,包括如下步骤:
步骤S1,提取:将卫矛科植物雷公藤的干燥根粉碎,乙醇溶液冷浸提取,回收提取液中的乙醇并用水稀释,过滤,收集滤液作为大孔树脂上样液;
步骤S2,XDA-1B大孔吸附树脂富集:以XDA-1B大孔吸附树脂为分离介质并装柱,树脂径高比为1:10,拌样树脂占树脂总量的1/10,湿法装柱,拌树脂上样;先用12BV的35%乙醇以12BV/h的流速清洗,后用8BV的75%乙醇(含万分之一三乙胺,体积百分含量)以12BV/h的流速洗脱并收集5-8BV洗脱液;将收集的洗脱液浓缩至干即得粗品;
步骤S3,碱开环酸闭环精制:将步骤S2所得粗品使用pH值为9.5的碱水溶解,过滤去除不溶物,滤液再用酸调节pH值至6.5,低温析晶,过滤收集析出物,洗涤、干燥,用65%乙醇(含万分之一三乙胺,体积百分含量)溶解作为上样液;
步骤S4,DM130大孔吸附树脂分离:以DM130大孔吸附树脂为分离介质并装柱,树脂径高比为1:20,拌样树脂占树脂总量的1/20,湿法装柱,拌树脂上样;用6BV的65%乙醇(含万分之一三乙胺,体积百分含量)以4BV/h的流速洗脱并收集5-6BV洗脱液,浓缩干燥即得雷公藤乙素和2-表雷公藤乙素混合物;
步骤S5,高速逆流色谱分离:以乙酸乙酯-正丁醇-水-冰醋酸(4:1:3:0.01)为溶剂体系,以其上相为固定相,下相为流动相;将步骤S4所得雷公藤乙素和2-表雷公藤乙素混合物用等体积固定相和流动相溶解,滤过,作为样品溶液;转速为850r/min,流动相流速为2.5mL/min,检测波长为220nm,根据色谱图收集雷公藤乙素、2-表雷公藤乙素色谱峰对应的洗脱液,浓缩干燥即得雷公藤乙素、2-表雷公藤乙素。
优选地,步骤S1提取的固液比为1:5,1kg干燥根对应用5L的乙醇溶液冷浸提取。
优选地,步骤S1用95%乙醇冷浸提取。
优选地,步骤S1回收提取液中的乙醇后用水稀释至0.5g生药/mL,过滤,收集滤液作为大孔树脂上样液。
优选地,步骤S2拌样树脂质量为上样液对应生药质量的1/2;步骤S4拌样树脂质量为上样液对应析出物质量的1/2。
优选地,步骤S3碱水为氨水溶液。
优选地,步骤S3酸为盐酸。
优选地,步骤S3低温析晶的温度为4℃。
优选地,步骤S3用65%乙醇(含万分之一三乙胺,体积百分含量)溶解成0.5g/mL溶液作为上样液。
优选地,步骤S5样品溶液的浓度为10mg/mL。
一种分离雷公藤乙素和2-表雷公藤乙素的高效液相色谱方法,以C18反相硅胶为固定相,以甲醇/乙腈/四氢呋喃/三乙胺/水=15/10/3/0.1/72(体积比)为流动相。
优选地,色谱柱为Ultimate XB-C18。
优选地,色谱柱填料粒径为5μm。
优选地,柱长250mm、内径4.6mm。
优选地,检测波长为220nm。
优选地,流动相流速为1.0mL/min。
优选地,进样量为10μL。
本发明的优点:
本发明提供的方法不依赖于反复硅胶柱层析即可分离制备雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素,适合于工业化生产。本发明提供的HPLC分析方法基于常规的C18色谱柱即可有效分离手性异构体雷公藤乙素和2-表雷公藤乙素,成本低、重复性高。
附图说明
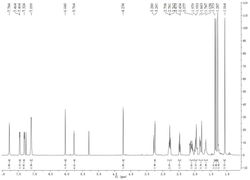
图1为XDA-1B大孔吸附树脂富集产物的HPLC检测色谱图,从该图可以看出,除了雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素,该产物还含有较多杂质;
图2为碱开环酸闭环精制产物的HPLC检测色谱图,从该图可看出,经过此步骤,雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素得到进一步富集,杂质成分显著减少;
图3为DM130大孔吸附树脂5-6BV洗脱液浓缩干燥产物的HPLC检测色谱图,基本只含有雷公藤乙素和2-表雷公藤乙素;
图4为DM130大孔吸附树脂8-9BV洗脱液浓缩干燥产物的HPLC检测色谱图,基本只含有雷公藤内酯酮;
图5为DM130大孔吸附树脂11-12BV洗脱液浓缩干燥产物的HPLC检测色谱图,基本只含有雷公藤甲素;
图6为使用不添加三乙胺的洗脱液洗脱的DM130大孔吸附树脂5-6BV洗脱液浓缩干燥产物的HPLC检测色谱图,雷公藤内酯酮与雷公藤乙素和2-表雷公藤乙素共洗脱;
图7为使用不添加三乙胺的洗脱液洗脱的DM130大孔吸附树脂8-9BV洗脱液浓缩干燥产物的HPLC检测色谱图,雷公藤内酯酮与雷公藤乙素和2-表雷公藤乙素共洗脱;
图8为HSCCC分离色谱图,雷公藤乙素和2-表雷公藤乙素基线分离;
图9为溶剂体系不含冰醋酸的HSCCC分离谱图,雷公藤乙素和2-表雷公藤乙素共洗脱;
图10为三种不同流动相对雷公藤乙素和2-表雷公藤乙素在C18色谱柱上的分离差异。
具体实施方式
下面结合附图和实施例具体介绍本发明实质性内容,但并不以此限定本发明的保护范围。
一、实验材料
雷公藤的干燥根购于亳州中药材市场,经鉴定为卫矛科植物雷公藤的干燥根;
XDA-1B大孔吸附树脂购于郑州勤实科技有限公司;
DM130大孔吸附树脂购于安徽三星树脂科技有限公司;
95%乙醇、三乙胺、乙酸乙酯、正丁醇、氨水和盐酸购于南京化学试剂股份有限公司;
TBE-300C型高速逆流色谱仪,上海同田生物技术有限公司。
二、实验方法
一种制备雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素的方法,包括:
步骤S1,提取:将卫矛科植物雷公藤的干燥根粉碎,95%乙醇冷浸过夜提取,固液比1:5,回收提取液中的乙醇并用水稀释至0.5g生药/mL,过滤,收集滤液作为大孔树脂上样液;
步骤S2,XDA-1B大孔吸附树脂富集:以XDA-1B大孔吸附树脂为分离介质并装柱,树脂径高比为1:10,拌样树脂占树脂总量的1/10,拌样树脂质量为上样液对应生药质量的1/2,湿法装柱,拌树脂上样;先用12BV的35%乙醇以12BV/h的流速清洗,后用8BV的75%乙醇(含万分之一三乙胺,体积百分含量)以12BV/h的流速洗脱并收集5-8BV洗脱液;将收集的洗脱液浓缩至干即得粗品;
步骤S3,碱开环酸闭环精制:将步骤S2所得粗品使用pH值为9.5的氨水溶解,过滤去除不溶物,滤液再用盐酸调节pH值至6.5,4℃低温析晶,过滤收集析出物,洗涤、干燥,用65%乙醇(含万分之一三乙胺,体积百分含量)溶解成0.5g/mL溶液作为上样液;
步骤S4,DM130大孔吸附树脂分离:以DM130大孔吸附树脂为分离介质并装柱,树脂径高比为1:20,拌样树脂占树脂总量的1/20,拌样树脂质量为上样液对应析出物质量的1/2,湿法装柱,拌树脂上样;用12BV的65%乙醇(含万分之一三乙胺,体积百分含量)以4BV/h的流速洗脱并收集5-6BV、8-9BV、11-12BV洗脱液;5-6BV洗脱液浓缩干燥即得雷公藤乙素和2-表雷公藤乙素混合物,8-9BV洗脱液浓缩干燥即得雷公藤内酯酮,11-12BV洗脱液浓缩干燥即得雷公藤甲素;
步骤S5,高速逆流色谱(HSCCC)分离:以乙酸乙酯-正丁醇-水-冰醋酸(4:1:3:0.01)为溶剂体系,以其上相为固定相,下相为流动相;将步骤S4所得雷公藤乙素和2-表雷公藤乙素混合物用10mL固定相和10mL流动相超声溶解,滤过,作为样品溶液;转速为850r/min,流动相流速为2.5mL/min,检测波长为220nm,根据色谱图收集雷公藤乙素、2-表雷公藤乙素色谱峰对应的洗脱液,浓缩干燥即得雷公藤乙素、2-表雷公藤乙素。
高速逆流色谱具体方法如下:
以乙酸乙酯-正丁醇-水-冰醋酸(4:1:3:0.01)混合液置于分液漏斗中,充分震荡后静置分层,分别取上下相,超声脱气20min。上相为固定相,下相为流动相。将固定相以20mL/min的体积流量将溶剂体系的上相注入HSCCC分离管中,待上相充满整个管路后调整主机转速为850r/min,再以2.5mL/min的体积流量注入流动相,两相在分离管中达到动态平衡后,由进样口注入20mL样品液(10mg/mL),温度25℃,检测波长220nm,得HSCCC分离图。
各样品的HPLC分析方法参数:
色谱柱:月旭Ultimate XB-C18(5μm,4.6×250mm)
流动相:甲醇/乙腈/四氢呋喃/三乙胺/水=15/10/3/0.1/72(体积比)等度洗脱
检测波长:220nm
流速:1.0mL/min。
三、实验结果
1、XDA-1B大孔吸附树脂富集结果
XDA-1B大孔吸附树脂的作用在于富集雷公藤中的雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素。富集的粗品的HPLC检测结果如图1所示,从图1可以看出,除了雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素,该粗品中还含有较多杂质。
2、碱开环酸闭环精制结果
碱开环酸闭环精制的步骤在于利用内酯类化合物在碱性条件下开环、酸性条件下闭环的原理进一步富集雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素。通过优化碱性条件和酸性条件的pH值可以最大限度地获得目标内酯类成分。该步骤精制产物的HPLC检测结果如图2所示,从图2可以看出,经过此步骤,雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素得到进一步富集,杂质成分显著减少。
3、DM130大孔吸附树脂分离结果
DM130大孔吸附树脂的作用在于分离雷公藤中的雷公藤内酯酮、雷公藤甲素、雷公藤乙素和2-表雷公藤乙素。5-6BV洗脱液浓缩干燥得到极性相对较大的雷公藤乙素和2-表雷公藤乙素混合物,HPLC检测结果如图3所示,二者总含量达到93.3%;8-9BV洗脱液浓缩干燥得到极性相对中等的雷公藤内酯酮,HPLC检测结果如图4所示,含量达到95.5%;11-12BV洗脱液浓缩干燥得到极性相对较小的雷公藤甲素,HPLC检测结果如图5所示,含量达97.1%。
经过多种不同类型的大孔吸附树脂和洗脱溶剂的优化,雷公藤乙素和2-表雷公藤乙素由于极性太相似均难以分离。雷公藤内酯酮在该条件下能与雷公藤乙素和2-表雷公藤乙素分离也是经过反复优化才实现的,比如若洗脱溶剂不添加三乙胺,5-6BV、8-9BV洗脱液所得产物的HPLC检测结果如图6、7所示,雷公藤内酯酮与雷公藤乙素和2-表雷公藤乙素共洗脱。
4、高速逆流色谱(HSCCC)分离结果
高速逆流色谱与大孔吸附树脂的分离原理不同,经过反复优化,雷公藤乙素和2-表雷公藤乙素在上述条件下得以分离,色谱图如图8所示。图9为溶剂体系中不添加冰醋酸的分离效果图,雷公藤乙素和2-表雷公藤乙素共洗脱。
5、HPLC分析方法的优化过程和结果
雷公藤乙素和2-表雷公藤乙素为一对手性异构体,化合物极性极为相似。对于这种手性异构体,通常采用手性色谱柱进行分离,常规的基于C18填料的反相色谱柱分离难度极大。但是,手性色谱柱极其昂贵,而且特别娇贵,待分析样品的杂质成分不能多,洗脱条件不能太剧烈,最不容忽视的缺陷是重复性差(尤其是不同实验室之间的重复性差)。使用手性色谱柱开发雷公藤乙素和2-表雷公藤乙素方法属于分析方法开发容易,应用稳定性差、成本高;使用反相色谱柱开发雷公藤乙素和2-表雷公藤乙素方法属于分析方法开发难,应用稳定性好、成本低。经过反复优化,得到上述HPLC分离参数,即:
色谱柱:月旭Ultimate XB-C18(5μm,4.6×250mm)
流动相:甲醇/乙腈/四氢呋喃/三乙胺/水=15/10/3/0.1/72(体积比)等度洗脱
检测波长:220nm
流速:1.0mL/min;
进样量:10μL;
液相色谱仪:Agilent 1260。
流动相采用的是三种有机溶剂混合,通过图10可以看出上述流动相体系的优势。样品溶液均为含有0.25mg/mL雷公藤乙素、0.25mg/mL 2-表雷公藤乙素的30%甲醇溶液。图10中A的流动相为甲醇/乙腈/四氢呋喃/三乙胺/水=15/10/3/0.1/72,B的流动相为甲醇/乙腈/三乙胺/水=20/10/0.1/70,C的流动相为甲醇/乙腈/三乙胺/水=14/14/0.1/72。图10中A、B、C色谱图使用的流动相极性相近,但对于雷公藤乙素和2-表雷公藤乙素在C18色谱柱上保留能力影响并不相同,甲醇/乙腈/四氢呋喃/三乙胺/水=15/10/3/0.1/72区分度好,雷公藤乙素和2-表雷公藤乙素在C18色谱柱上达基线分离,该流动相可用于分离分析雷公藤乙素和2-表雷公藤乙素。
上述实施例的作用在于具体介绍本发明的实质性内容,但本领域技术人员应当知道,不应将本发明的保护范围局限于该具体实施例。
一种制备雷公藤甲素的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0