






专利摘要
本发明涉及一种冠醚类化合物,所述的冠醚类化合物,其为式I所示化合物、或其异构体。本发明提供的冠醚类化合物不仅可与铵盐形成配合物,而且在光照和/或加热后,铵盐分子可与所述冠醚类化合物分离。如此,不仅丰富了冠醚的种类,而且拓宽了冠醚的应用领域。式I中,R1~R6分别独立选自:C1~C3直链或直链的烷基,n为大于或等于5的整数。
权利要求
1.一种冠醚化合物,其为式I所示化合物:
式I中,R
2.如权利要求1所述的冠醚化合物,其特征在于,其中R
3.一种制备权利要求1所述的冠醚化合物的方法,其特征在于,所述方法包括如下步骤:
(1)由式IIa所示化合物和式IIIa所示化合物反应,得到式IVa所示化合物的步骤;或,式IIb所示化合物和式IIIb所示化合物反应,得到式IVb所示化合物的步骤;
(2)式IVa所示化合物溴代,得到式Va所示化合物的步骤;或,式IVb所示化合物溴代,得到式Vb所示化合物;
(3)式Va所示化合物与式Vb所示化合物经偶联反应,得到式VI所示化合物;
(4)由式VI所示化合物制备式VII所示化合物的步骤;
(5)式VII所示化合物制备式VIII所示化合物的步骤;和
(6)式VIII所示化合物与式IX所示化合物反应,制备目标物的步骤;
其中,R
4.如权利要求3所述的方法,其特征在于,其中所述步骤(1)的主要步骤是:由式IIa所示化合物或式IIb所示化合物和式IIIa所示化合物或式IIIb所示化合物于多聚磷酸中反应,反应温度为80℃~120℃,得到式IVa所示化合物或式IVb所示化合物。
5.如权利要求3所述的方法,其特征在于,其中所述步骤(2)的主要步骤是:由式IVa所示化合物或式IVb所示化合物与N-溴代丁二酰亚胺与由乙腈和15%硫酸组成的混合物中在70℃~85℃反应,其中,式IVa所示化合物或式IVb所示化合物与N-溴代丁二酰亚胺的摩尔比为1:(1~1.5)。
6.如权利要求3所述的方法,其特征在于,其中所述步骤(3)的主要步骤是:由式Va所示化合物与式Vb所示化合物在锌粉和四氯化钛作用下进行McMurry偶联反应,其中四氯化钛和锌粉摩尔比为1:(2~3),反应溶剂为四氢呋喃,反应温度为70℃~85℃。
7.如权利要求3所述的方法,其特征在于,其中所述步骤(4)的主要步骤是:在有钯催化剂和碱存在条件下,由式VI所示化合物与联硼酸频那醇酯在1,4-二氧六环中于90℃~120℃反应,得到式VII所示化合物。
8.如权利要求3所述的方法,其特征在于,其中所述步骤(5)的主要步骤是:在碱性条件下,由式VII所示化合物与双氧水在由10%氢氧化钠水溶液和四氢呋喃组成的化合物中,于20℃~30℃反应,得到式VIII所示化合物。
9.如权利要求3所述的方法,其特征在于,其中所述步骤(6)的主要步骤是:在有碱存在条件下,将式VIII所示化合物与式IX所示化合物置于乙腈中,于70℃~85℃反应,得到目标物。
说明书
技术领域
本发明涉及一种冠醚类化合物及其制备方法;具体说,涉及一种含芳基烯烃基团的冠醚类化合物及其制备方法。
背景技术
冠醚是一种包含有多个醚基团的杂环有机化合物,其能络合碱金属离子(C.J.Pedersen,J.Am.Chem.Soc.,1967,89,7017)。现有冠醚除了可络合钾、钠等碱金属离子外,还可以作为主体分子,通过氢键与铵盐(客体分子)形成配合物。
然而,现有冠醚(如苯并24冠8等)一旦与铵盐形成配合物,较难再将铵盐分子脱离(包括采用光照和/或加热等方法)。如此,限制了冠醚的应用领域。
发明内容
本发明的发明人经广泛且深入的研究,设计并合成了一种结构新颖的冠醚类化合物。该类冠醚类化合物不仅可与铵盐形成配合物,而且在光照和/或加热后,铵盐分子可与所述冠醚类化合物分离。如此,不仅丰富了冠醚的种类,而且拓宽了冠醚的应用领域(本发明所述冠醚类化合物可用于分子识别和药物载体等领域)。
本发明的一个目的在于,提供一种结构新颖的冠醚类化合物。
本发明所述的冠醚类化合物,其为式I所示化合物、或其异构体(包括立体异构或顺反异构):
式I中,R1~R6分别独立选自:C1~C3直链或直链的烷基,n为大于或等于5的整数。
本发明另一个目的在于,提供一种制备式I所示化合物的方法。
所述方法包括如下步骤:
(1)由式IIa所示化合物和式IIIa所示化合物反应(或式IIb所示化合物和式IIIb所示化合物反应),得到式IVa所示化合物(或式IVb所示化合物)的步骤;
(2)式IVa所示化合物(或式IVb所示化合物)溴代,得到式Va所示化合物(或式Vb所示化合物)的步骤;
(3)式Va所示化合物与式Vb所示化合物经偶联反应,得到式VI所示化合物的步骤的步骤;
(4)由式VI所示化合物制备式VII所示化合物的步骤;
(5)式VII所示化合物制备式VIII所示化合物的步骤;和
(6)式VIII所示化合物与式IX所示化合物反应,制备目标物(式I所示化合物)的步骤。
其中,R1~R6和n的含义与前文所述相同,式IX所示化合物可由式X所示化合物与对甲基苯磺酰氯反应制得,具体可参见文献(J.Med.Chem.2016,59,17,7840-7855)。
附图说明
图1.为式I-1所示化合物核磁共振氢谱;
图2.为式I-2所示化合物核磁共振氢谱;
图3.为式I-1所示化合物及式I-1所示化合物与二苄铵六氟磷酸正离子以摩尔比为1:1混合所得混合物的核磁共振氢谱(氘代氯仿)对比图;
其中,A为式I-1所示化合物的核磁共振氢谱,B为式I-1所示化合物与二苄铵六氟磷酸正离子(摩尔比为1:1混合)混合物核磁共振氢谱;
图4.为式I-2所示化合物及式I-2所示化合物与二苄铵六氟磷酸正离子以摩尔比为1:1混合所得混合物的核磁共振氢谱(氘代乙腈)对比图;
其中,A为式I-2所示化合物的核磁共振氢谱,B为式I-2所示化合物与二苄铵六氟磷酸正离子(摩尔比为1:1混合)混合物核磁共振氢谱;
图5.为式I-1所示化合物与二苄铵六氟磷酸正离子以摩尔比为1:1混合所得混合物在光照及加热前、后核磁共振氢谱(氘代乙腈)对比图;
其中,A为所述混合物未经光照及加热的核磁共振氢谱,B为所述混合物经光照及加热的核磁共振氢谱;
图6.为式I-2所示化合物与与二苄铵六氟磷酸正离子以摩尔比为1:1混合所得混合物在光照及加热前、后核磁共振氢谱(氘代乙腈)对比图;
其中,A为所述混合物未经光照及加热的核磁共振氢谱,B为为所述混合物经光照及加热的核磁共振氢谱;
图7.为苯并24冠8(简记为:“DB24C8”,下同)、由DB24C8与二苄铵六氟磷酸正离子以摩尔比为1:1组成的混合物在光照(310nm)和加热(65℃)前、后的核磁共振氢谱(氘代乙腈)对比图;
其中,A为DB24C8的核磁共振氢谱,B为DB24C8与二苄铵六氟磷酸正离子以摩尔比为1:1组成的混合物未经光照(310nm)和加热(65℃)的核磁共振氢谱,C为DB24C8与二苄铵六氟磷酸正离子以摩尔比1:1组成的混合物经光照(310nm)和加热(65℃)后的核磁共振氢谱。
具体实施方式
在本发明一个优选的技术方案中,R1~R6分别独立选自:甲基,乙基或丙基中一种;
在一个更优选的技术方案中,R1~R6均为甲基。
在本发明另一个优选的技术方案中,n为5~7的整数。
本发明提供的制备式I所示化合物的方法,具体包括如下步骤:
(1)式IIa所示化合物(或式IIb所示化合物)和式IIIa所示化合物(或式IIIb所示化合物)于多聚磷酸中反应,反应温度为80℃~120℃,式IIa所示化合物(或式IIb所示化合物)与式IIIa所示化合物(或式IIIb所示化合物)的摩尔比为1:(1~1.5),得到式IVa所示化合物(或式IVb所示化合物);
(2)式IVa所示化合物(或式IVb所示化合物)与N-溴代丁二酰亚胺于由乙腈和15%硫酸组成的混合物中在70℃~85℃反应,其中,式IVa所示化合物(或式IVb所示化合物)与N-溴代丁二酰亚胺的摩尔比为1:(1~1.5),得到式Va所示化合物(或式Vb所示化合物);
(3)式Va所示化合物与式Vb所示化合物在锌粉和四氯化钛作用下进行McMurry偶联反应,其中四氯化钛和锌粉摩尔比为1:(2~3),反应溶剂为四氢呋喃,反应温度为70℃~85℃,得到式VI所示化合物;
(4)在有钯催化剂和碱存在条件下,由式VI所示化合物与联硼酸频那醇酯在1,4-二氧六环中于90℃~120℃反应,得到式VII所示化合物;
其中,所述钯催化剂优选为[1,1'-双(二苯基膦基)二茂铁]二氯化钯,所述的碱优选为醋酸钾,式VI所示化合物与联硼酸频那醇酯的摩尔比优选为1:(2.2~2.5);
(5)在碱性条件下,由式VII所示化合物与双氧水在由10%氢氧化钠水溶液和四氢呋喃组成的化合物中,于20℃~30℃反应,得到式VIII所示化合物;
其中,式VII所示化合物与双氧水的摩尔比是1:(2~3);和,
(6)在有碱存在条件下,将式VIII所示化合物与式IX所示化合物置于乙腈中,于70℃~85℃反应,得到目标物(式I所示化合物);
其中,所述的碱优选为碳酸钾,式VIII所示化合物与式IX所示化合物的摩尔比为1:1,制备式IX所示化合物的主要步骤是:在有有机碱(如三乙胺等)存在条件下,由式X所示化合物与对甲基苯磺酰氯在四氢呋喃于0℃~25℃反应,得到式IX所示化合物,式X所示化合物与对甲基苯磺酰氯的摩尔比为1:(4~5)。
下面通过实施例对本发明作进一步说明。其目的仅在于更好理解本发明的内容。因此,所举之例不限制本发明的保护范围。
实施例1
式I-1所示化合物的合成:
(1)2,4,7-三甲基-1-二氢茚酮(式IVa-1所示化合物)的合成:
将对二甲苯(40g,0.377mol)置于反应瓶中,加入500g多聚磷酸,用电动搅拌是对二甲苯和多聚磷酸混合均匀,滴加2-甲基丙烯酸(32.4g,0.377mol),100℃加热两小时。薄层色谱(TLC)跟踪监测反应,至原料耗尽。加水,用乙酸乙酯萃取三次,有机相合并用无水硫酸钠干燥,旋干,柱层析分离(洗脱剂为:石油醚:乙酸乙酯=300:1。v/v),得到25g浅黄色油状液体(式IVa-1所示化合物),产率38%。
(2)2,4,7-三甲基-6-溴-1-二氢茚酮(式Va-1所示化合物)的合成:
将式IVa-1所示化合物(25g,0.143mol)溶于300ml乙腈中,加入15%硫酸100ml和N-溴代丁二酰亚胺(33.2g,0.187mol),80℃回流14h,,TLC跟踪监测反应,至原料耗尽。旋干乙腈,并用乙酸乙酯萃取三次,合并有机层用无水硫酸钠干燥,旋干,柱层析分离(洗脱剂为石油醚:乙酸乙酯=100:1,v/v),得到10g浅黄色固体(式Va-1所示化合物),产率27.6%。
(3)式VI-1所示化合物的合成:
取锌粉(10.27g,0.158mol)加入300ml无水四氢呋喃,在冰浴条件下,滴加四氯化钛(14.9g,0.079mol),在75℃回流2小时
将式Va-1所示化合物(10g,0.0395mol)溶于四氢呋喃(THF)中,缓慢加入上述体系。75℃环流48h,TLC跟踪监测反应,至原料耗尽。再加入饱和氯化铵100ml,用乙酸乙酯萃取三次,合并有机层,并用无水硫酸钠干燥,旋干,柱层析分离(洗脱剂为:石油醚:乙酸乙酯=500:1,v/v),得到白色固体,甲醇二氯甲烷重结晶,得7.9g的式VI-1所示化合物,产率85%。
(4)式VII-1所示化合物的合成:
将式VI-1所示化合物(7.9g,0.0166mol),联硼酸频那醇酯(10.2g,0.04mol)溶于1,4-二氧六环(200ml)中;
将无水醋酸钾(9.79g,0.1mol)和[1,1'-双(二苯基膦基)二茂铁]二氯化钯(2.44g,0.0033mol),加入到上述体系中,110℃回流2小时,TLC跟踪监测反应至原料耗尽。抽滤,旋蒸蒸除1,4-二氧六环,用饱和食盐水洗,二氯甲烷萃取三次,有机相合并后用无水硫酸钠干燥,旋干,柱层析分离(洗脱剂为:石油醚:乙酸乙酯=100:1,v/v),得到7.09g浅黄色粉末(式VII-1所示化合物),收率75%。
(5)式VIII-1所示化合物的合成:
将式VII-1所示化合物(7.09g,0.0125mol)溶于四氢呋喃(100ml)中,加入30ml的10%氢氧化钠水溶液,再加入30ml的10%双氧水常温搅拌10分钟,TLC跟踪监测反应至原料耗尽,加入硫代硫酸钠,用乙酸乙酯萃取三次,合并有机层,用无水硫酸钠干燥,旋干,柱层析分离(洗脱剂为:石油醚:乙酸乙酯=50:1,v/v),得到3.5g浅白色粉末(式VIII-1所示化合物),得率80%。
(6)目标物的合成:
将式X-1所示化合物(1g,3.54mmol)溶于四氢呋喃(50ml)中,加入三乙胺(1.79g,17.7mmol),冰浴下滴加对甲苯磺酸酰氯(3.04g,15.9mmol),滴加完后冰浴搅拌30min,再常温搅拌5h,TLC跟踪监测反应至原料耗尽,过滤滤去固体,旋干四氢呋喃,加入100ml水,用乙酸乙酯萃取三次,合并有机层,用无水硫酸钠干燥,旋干,柱层析分离(洗脱剂为石油醚:乙酸乙酯=20:1,v/v),得到1.89g无色液体(式IX-1所示化合物),得率90%;
将式VIII-1所示化合物(300mg,0.862mmol)和式IX-1所示化合物(509mg,0.862mmol)溶于300ml乙腈中,加入碳酸钾(714mg,5.17mmol),75℃回流168h,TLC跟踪监测反应至原料耗尽,过滤滤去固体,旋干乙腈,加30ml去离子水,用二氯甲烷萃取三次,合并有机层,用无水硫酸钠干燥,旋干,柱层析分离(洗脱剂为:石油醚:乙酸乙酯=1:1,v/v),得到120mg浅白色固体(式I-1所示化合物,目标化合物),得率25%。
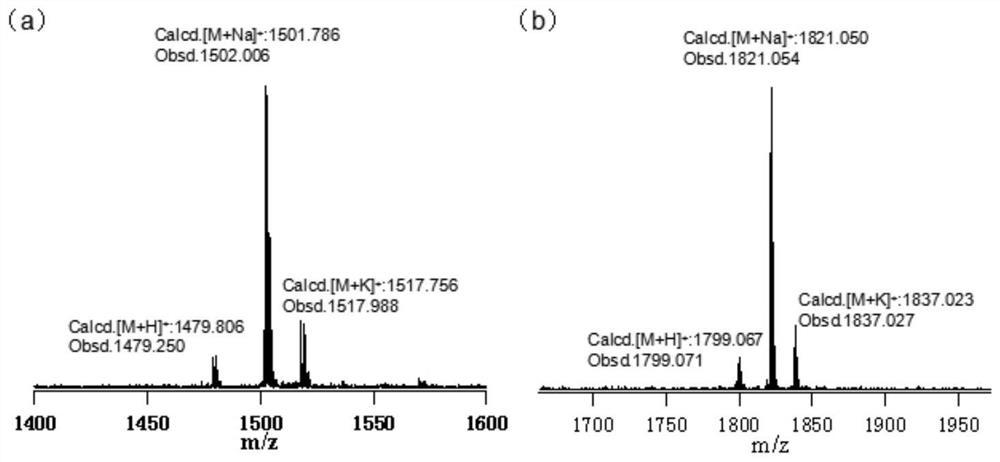
HRMS(ESI):FormulaCalc.Mass[M+Na
实施例2
式I-2所示化合物的合成:
除了以式X-2所示化合物替换实施例1中式X-1所示化合物外,其它步骤及所用试剂与实施例1相同。得到75mg浅白色固体(式I-2所示化合物,目标化合物),得率25%。
HRMS(ESI):FormulaCalc.Mass[M+Na
实施例3
(1)将式I-1所示化合物(主体分子)与二苄铵六氟磷酸正离子(客体分子)以摩尔比为1:1混合得化合物,然后在氘代氯仿中检测所得混合物的核磁共振氢谱,并与式I-1所示化合物核磁共振氢谱(氘代氯仿)对比,具体见图3(图3中,A为主体分子的核磁共振氢谱,B为混合物的核磁共振氢谱)。
同样,将式I-2所示化合物(主体分子)与二苄铵六氟磷酸正离子(客体分子)以摩尔比为1:1混合得化合物,然后在氘代乙腈中检测所得混合物的核磁共振氢谱,并与式I-2所示化合物核磁共振氢谱(氘代乙腈)对比,具体见图4(图4中,A为主体分子的核磁共振氢谱,B为混合物的核磁共振氢谱)。
由图3(或图4)可知:主客体分子相互结合后,图3(或图4)中,烷氧链部分(即A1和B1对比)显示氢谱变宽变矮,芳香氢A2和B2发生化学位移。据此说明式I-1所示化合物(或式I-2所示化合物)与二苄铵六氟磷酸正离子结合为了主客体分子。
(2)将式I-1所示化合物(或式I-2所示化合物)与二苄铵六氟磷酸正离子以摩尔比1:1混合得混合物,分别检测所得混合物在光照(310nm)及加热(65℃)前、后的核磁共振氢谱(氘代乙腈)结果分别见图5(或图6)(图5(或图6)中,A为所述混合物未经光照及加热的核磁共振氢谱,B为所述混合物经光照及加热的核磁共振氢谱)。
由图5(或图6)可知,A2A3对应B2B3核磁部分显示光照后冠醚(式I-1或I-2所示化合物,下同)中芳基乙烯部分由CIS变成Trans构型,并且低场区的二苄铵六氟磷酸正离子上的芳香氢由原来的宽矮峰(A1)变成了尖高峰(B1),这些证据显示,二苄铵六氟磷酸正离子(客体分子)随着冠醚中芳基乙烯部分的转动,与冠醚的结合能力减弱,从冠醚中脱离出来。
对比实施例
分别检测DB24C8(苯并24冠8)、由DB24C8与二苄铵六氟磷酸正离子以摩尔比为1:1组成的混合物在光照310nm和65℃加热前、后的核磁共振氢谱(氘代乙腈),结果见图7。
由图7可知,B1B2相对于A1A2在高场都产生了新的峰,显示DB24C8与二苄铵六氟磷酸正离子结合,再由C1C2和B1B2对比发现光照后及加热后体系没有发生变化,说明在光照加热情况下,对DB24C8(现有冠醚)与对二苄铵六氟磷酸正离子结合力没有影响,换而言之,二苄铵六氟磷酸正离子在光照和加热后没有和DB24C8脱离。
新型冠醚类化合物及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




















动态评分
0.0