

IPC分类号 : C08G77/42,C08G77/60,C08G77/38,C08G77/28,C08G77/04








专利摘要
本发明公开了一种硅基超支化共轭聚合物及其制备方法,该超支化聚合物是由若干个支化单元聚合而成,其数均分子量为3~6×104g·mol‑1,分子量分布指数1.05~1.50,每个支化单元是以八(3‑巯丙基)‑三维笼型倍半硅氧烷(POSS)为核心化合物,所述核心化合物的末端巯基连接聚‑(二卤甲基苯基硅烷‑co‑二卤二苯基硅烷)。该超支化聚合物的特点在于:1)具有三维孔道结构,增加硝基芳烃的渗透性与比表面积;2)正电性(δ+)的硅原子与硝基芳烃的氮原子或氧原子之间的弱非共价相互作用,可有效地富集硝基芳烃分子。
权利要求
1.一种硅基超支化共轭聚合物,该超支化聚合物是由若干个支化单元聚合而成,其特征是:该聚合物的数均分子量为3~6×10
所述聚(甲基苯基硅烷-co-二苯基硅 烷)为无规共聚硅烷;
形成聚(甲基苯基硅烷-co-二苯基硅烷)的原料二卤甲基苯基硅烷和二卤二苯基硅烷的摩尔比例为1.0:0.5~1.5。
2.权利要求1所述的硅基超支化共轭聚合物的制备方法,其特征是,包括以下步骤:
将八(3-巯丙基)-三维笼型倍半硅氧烷和聚(甲基苯基硅烷-co-二苯基硅烷)通过巯基-烯点击化学反应合成所述的硅基超支化共轭聚合物。
3.如权利要求2所述的制备方法,其特征是:
聚(甲基苯基硅烷-co-二苯基硅烷)的合成,包括以下步骤:利用伍尔兹反应,在钠沙甲苯分散体系中,将二卤甲基苯基硅烷和二卤二苯基硅烷共聚,回流;然后加入甲醇并收集粗产物,依次用甲苯,四氢呋喃和水洗涤,得到聚(甲基苯基硅烷-co-二苯基硅烷)。
4.如权利要求1所述的制备方法,其特征是:形成聚(甲基苯基硅烷-co-二苯基硅烷)的原料二卤甲基苯基硅烷和二卤二苯基硅烷的摩尔比例为1.0:1.3。
5.如权利要求2所述的制备方法,其特征是:所述巯基-烯点击化学反应中采用的反应溶剂为无水甲苯。
6.如权利要求2所述的制备方法,其特征是:所述的巯基-烯点击化学反应中采用的自由基引发剂为偶氮二异丁腈。
7.如权利要求2所述的制备方法,其特征是:合成硅基超支化共轭聚合物的反应时间为4~8小时;反应温度为60~80℃。
8.如权利要求7所述的制备方法,其特征是:合成硅基超支化共轭聚合物的反应时间为4小时;反应温度为60℃。
9.如权利要求2所述的制备方法,其特征是:合成硅基超支化共轭聚合物的反应在惰性气体条件下进行,所述惰性气体为氩气或氮气。
10.如权利要求2所述的制备方法,其特征是:巯基-烯点击化学反应反应时,采用乙烯基聚(甲基苯基硅烷-co-二苯基硅烷);八(3-巯丙基)-三维笼型倍半硅氧烷、乙烯基聚(甲基苯基硅烷-co-二苯基硅烷)和偶氮二异丁腈的质量比例为1.0:0.1~0.6:0.10~0.15;
乙烯基聚(甲基苯基硅烷-co-二苯基硅烷)的合成方法,包括以下步骤:室温下,将乙烯基卤化镁加入到聚(甲基苯基硅烷-co-二苯基硅烷)无水THF溶液中;并将溶液加热搅拌;通过蒸馏除去THF并浓缩产物;将剩余的反应混合物溶于二氯甲烷中,过滤除去副产物盐;最终,得到乙烯基聚(甲基苯基硅烷-co-二苯基硅烷);
合成乙烯基聚(甲基苯基硅烷-co-二苯基硅烷)的反应温度为40~60℃;所述乙烯基卤化镁与聚(甲基苯基硅烷-co-二苯基硅烷)的添加比例为0.1~1mL:0.1~0.15g。
11.如权利要求10所述的制备方法,其特征是:八(3-巯丙基)-三维笼型倍半硅氧烷、乙烯基聚(甲基苯基硅烷-co-二苯基硅烷)和偶氮二异丁腈的质量比例为1:0.6:0.10。
12.如权利要求10所述的制备方法,其特征是:合成乙烯基聚(甲基苯基硅烷-co-二苯基硅烷)的反应温度为60℃;所述乙烯基卤化镁与聚(甲基苯基硅烷-co-二苯基硅烷)的添加比例为0.1mL:0.13g。
13.如权利要求2所述的制备方法,其特征是:具体反应过程包括:在惰性气体环境中,将八(3-巯丙基)-三维笼型倍半硅氧烷、偶氮二异丁腈和乙烯基聚(甲基苯基硅烷-co-二苯基硅烷)混合,油浴加热;然后除去油浴,将溶液暴露于空气中;沉淀回收产物,用氯仿洗涤,合并滤液在室温下静置8h~16h,干燥,得到所述的硅基超支化共轭聚合物。
说明书
技术领域
本发明属于聚合物合成和荧光传感材料技术领域,具体涉及一种以笼型倍半硅氧烷(POSS)为核心,以聚硅烷为侧链的硅基超支化共轭聚合物及其制备方法。
背景技术
爆炸物的在线检测主要是对爆炸物挥发出的蒸气和对粘附于爆炸物容器表面以及任何接触过爆炸物的物件(包括人体)表面所残留的微痕量爆炸物进行检测的技术。隐藏性爆炸物会在其周围一定范围内形成微痕量的浓度。据报道,PVC外壳的埋地地雷经过一段时间后在地表中的TNT、DNT含量可达0.15ppb。而荧光传感法具有灵敏度高、可采集参数多(如荧光强度、荧光光谱形貌、荧光各向异性、荧光寿命等)、响应时间快及仪器设计相对成熟等特点成为最具有开发前景的探测方法。
目前已经有大量用于检测NACs爆炸物的荧光传感材料被报道,但大多数是有机π-π共轭聚合物(OCP)。OCP的骨架可以作为分子导线,使激子能够沿着聚合物链快速迁移,从而产生“一点接触、多点响应”的信号放大效应。然而,在固体膜中,OCP倾向于通过聚合物主链之间的π-π相互作用堆集。这种π–π堆集导致两个缺点,限制了它们在化学传感器中的应用:1)由聚合物主链之间的能量迁移引起的荧光自猝灭现象;2)OCP的主链往往为刚性的,导致其在常用溶剂中的溶解性较差,难以加工利用。
聚硅烷是近年来出现的一种新的无机共轭高分子材料。其主链由Si—Si组成,与由C=C及芳环构成的刚性共轭聚合物在性质上差别明显。首先,Si—Si键构成的分子主链为柔性链,溶解性好,易于加工;其次,Si原子较小的电负性以及空的3d轨道,使得聚硅烷主链的σ电子能够沿着Si—Si主链广泛离域,产生σ共轭效应。因此与OCP类似,聚硅烷也具有分子导线效应,对硝基芳烃爆炸物具有较好的响应灵敏度。但聚硅烷的柔性主链使其避免了类似OCP中π–π堆积产生的荧光猝灭。再次,正电性(δ+)的硅原子与硝基芳烃中负电性的氧原子之间的弱非共价相互作用,可有效地富集硝基芳烃分子,提高响应灵敏度。总之聚硅烷是一种很有潜力的检测硝基芳烃的荧光传感材料。然而单纯的聚硅烷作为荧光传感材料,其分子链之间缠结紧密,渗透性较差,不利于待测物分子在其中的扩散。
多面体低聚倍半硅氧烷(POSS)作为一种纳米笼状分子,在每个硅原子顶点可用有机基团修饰。由于其独特的纳米尺寸结构、较高的热稳定性、以及容易化学修饰等特点,POSS已被证明是一种重要的3D支架,用于各种用途的多孔材料的构建。若将其引入荧光传感分子中,可大大提高材料的渗透性,提高传感灵敏度。目前已有多种以POSS为核心的化合物的合成方法及光学性能的报道。这类化合物呈现三维立体结构,可在分子内部产生微小孔隙,材料比表面大,分子渗透性好,有利于提高传感材料的灵敏度。
因此,以POSS为核心、以聚硅烷为侧链构筑一种用于TNT、DNT的荧光分析检测的超支化共轭聚合物,既具有聚硅烷优良的传感性能,又具有POSS衍生物的高渗透性,因而会表现出较好的综合性能。
发明内容
针对以上现有技术,本发明的目的是提供一种以POSS为核心、以聚硅烷为侧链的超支化荧光聚合物(化合物IV)。其中聚硅烷为σ共轭结构,具有分子导线效应,无π–π堆积而导致的荧光猝灭;三维的POSS结构提高待测物分子在膜中的渗透速率,从而能提高材料的传感灵敏度。
本发明采用以下技术方案:
本发明的第一个方面,提供了一种硅基超支化共轭聚合物,该超支化聚合物是由若干个支化单元聚合而成,其数均分子量为3~6×10
该超支化聚合物具有微孔和介孔结构,其中,介孔孔径分布在2~55nm之间,主要孔径分布在15~20nm(优选17nm)。该聚合物具有丰富的孔道结构,增加了其比表面,提高了硝基芳烃的渗透速率,检测灵敏度提高。
进一步的,本发明聚合物所含的聚-(二卤甲基苯基硅烷-co-二卤二苯基硅烷)嵌段,为无规聚合物。经过实验验证,该聚硅烷对硝基芳烃表现出了良好的荧光猝灭性能,且在普通溶剂中有很好的溶解性。
更进一步的,聚-(二卤甲基苯基硅烷-co-二卤二苯基硅烷)为聚-(二氯甲基苯基硅烷-co-二氯二苯基硅烷)或聚-(二溴甲基苯基硅烷-co-二溴二苯基硅烷)。
本发明的第二个方面,还提供上述硅基超支化共轭聚合物的制备方法,包括以下步骤:
将八(3-巯丙基)POSS和聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)通过巯基-烯点击化学反应合成所述的硅基超支化共轭聚合物。
本发明中,所述巯基-烯点击化学反应中采用的反应溶剂为无水甲苯。
本发明中,所述的巯基-烯点击化学反应中采用的自由基引发剂为偶氮二异丁腈(AIBN)。
本发明中,合成硅基超支化共轭聚合物的反应时间为4~8小时,进一步优选的反应时间为4小时;反应温度为60~80℃,进一步优选的反应温度为60℃。
本发明中,合成硅基超支化共轭聚合物的反应在惰性气体条件下进行,所述惰性气体可为氩气或氮气。在本发明中,所述八(3-巯丙基)POSS、乙烯基共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)和偶氮二异丁腈(AIBN)的质量比例为1.0:0.1~0.6:0.10~0.15,进一步优选的质量比例为1:0.6:0.10。
本发明中,一些优选的实施方式中,具体包括以下步骤:
在N2环境中,将八(3-巯丙基)POSS、AIBN和乙烯基聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)混合,油浴加热;然后除去油浴,将溶液暴露于空气中;回收沉淀产物,用氯仿洗涤,合并滤液在室温下静置过夜(8h~16h),干燥,得到白色粉末状化合物IV。
相比于现有技术,本发明的技术方案具有如下有益效果:
本发明制备的化合物与目前所报道的以笼型倍半硅氧烷POSS为核心的化合物相比,独特之处在于在各个顶点功能化地引入具有σ共轭效应的聚硅氧烷发色团。该超支化聚合物的特点在于:1)具有三维孔道结构,增加硝基芳烃的渗透性与接触面积;2)正电性(δ
附图说明
图1为本发明化合物的合成路线图。
图2为本发明化合物I、化合物II、化合物IV的红外波谱对比。
图3为本发明化合物IV的X射线衍射的测试图。
图4为本发明化合物IV固体颗粒的SEM图片。
图5为本发明化合物的氮气吸脱附(A)及其孔径分布(B)曲线。
图6为本发明化合物II和IV及其旋涂膜玻璃片的紫外可见吸收(A)与荧光发射(B)光谱。
图7为化合物II(A)和IV(B)对TNT的THF溶液的荧光猝灭图。
图8为化合物II和IV对TNT的THF溶液的猝灭率曲线(A)和KSV(B)曲线图。
图9为在DNT饱和蒸汽中化合物II(A)和化合物IV(B)旋涂膜的荧光随时间的变化图。
图10为化合物II和IV的旋涂膜的猝灭率曲线对比图。
图11为化合物IV制作的便携式试对TNT溶液的猝灭效果图。
图12为化合物IV旋涂膜的荧光猝灭可逆性效果图。
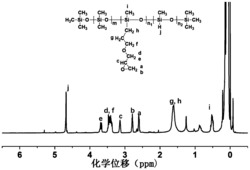
图13为八(3-巯丙基)POSS(I)核磁共振氢谱图。
图14为聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(II)核磁谱图。
图15为三维超支化共轭聚合物(3D-HPs)(IV)核磁谱图。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作和/或它们的组合。
本发明中的实验原料试剂如表1。
表1主要原料及试剂
本发明实验仪器设备如表2。
表2主要实验仪器设备
本发明中的术语解释:
本发明中过夜是指8~16h。
本发明中若干个是指大于等于两个以上。
本发明中室温是指18~37℃。
本发明中的σ共轭是指在聚硅烷中,由于Si原子较小的电负性以及空的3d轨道,使得聚硅烷主链的σ电子能够沿着Si—Si主链广泛离域,产生σ共轭效应。
正如背景技术中所介绍的,针对现有技术中的不足,为了解决如上的技术问题,本发明提出了一种硅基超支化共轭聚合物,该超支化聚合物是由若干个支化单元聚合而成,其数均分子量为3~6×10
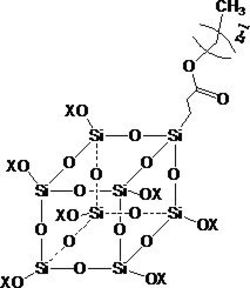
其中,所述八(3-巯丙基)-三维笼型倍半硅氧烷(POSS)是指T8型三维笼型倍半硅氧烷的八个顶角硅原子连接巯丙基,末端基团是巯基。
三维笼型倍半硅氧烷,也称多面体低聚倍半硅氧烷,英文名称polyhedraloligomeric silsesquioxane,简称POSS,它是一种低聚硅氧烷,在纳米级尺度上具有笼型结构的特点。通式(RSiO1.5)n,式中n为6、8、10或12等,可以进一步简称为T6、T8、T10或T12等。从制备的容易程度及对提高材料渗透性的效果来讲,本发明经过优化选择n为8的笼型倍半硅氧烷(POSS)作为超支化聚合物的核心。
在本发明中,由两种或两种以上单体共同参加的聚合反应,称做共聚合,所形成的聚合物含有两种或两种以上单体单元,这类聚合物称做共聚物,可分为无规共聚物、交替共聚物、嵌段共聚物和接枝共聚物。由于共聚物包含至少两种结构单元,它可以根据其结构单元的排列顺序分成四种共聚物,当结构单元为A和B时的情况共聚物的命名是将两单体之间加连接号,然后用括号括起,前面加上聚字,比如聚(二氯甲基苯基硅烷-二氯二苯基硅烷)。国际命名中常在两单体名间加入alt、co、b、g和分别代表交替、无规、嵌段和接枝共聚物,比如聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)或共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷),即表示二卤甲基苯基硅烷-二卤二苯基硅烷的无规共聚物。
本发明中的一个具有代表性的超支化聚合物的结构如1所示,其相应的支化单元的结构如2所示。
其中,我们可以看到聚-(二卤甲基苯基硅烷-co-二卤二苯基硅烷)采用了如1和2所示的结构表示,这种共聚物结构的表示方法是在共聚技术领域中的一种常规的表示方法,这里对共聚物结构的解读是:(1)它可以代表无规共聚物,共聚物中两结构单元随机出现,并没有一定的规律,但是共聚物中的二卤二苯基硅烷单体的总个数是n,二卤甲基苯基硅烷的总个数为m,对于在式1或式2所示的结构中,巯基连接的是二卤甲基苯基硅烷或者二卤二苯基硅烷,而并不是仅仅指巯基与二卤二苯基硅烷直接相连,这种结构的表示是由于无规共聚的特殊性决定的,但是经过以上解释,本领域技术人员可常规知晓其结构含义;(2)它可以代表交替共聚物,表示的是两种单体(二卤甲基苯基硅烷和二卤二苯基硅烷)在大分子链上严格相间排列,此时n等于m,或n±1等于m,巯基连接的是二卤甲基苯基硅烷或者二卤二苯基硅烷,而并不是仅仅指巯基与二卤二苯基硅烷直接相连;(3)它可以代表嵌段共聚物,是将两种性质不同的聚合物链段连在一起制备而成的一种特殊聚合物,也就是指n个二卤二苯基硅烷单体所形成的聚合物链段与m个二卤甲基苯基硅烷所形成的聚合物链段在一起制备而成的聚合物。
其中,m为50~60,n为35~45,波浪线代表通过巯丙基连接的其它各顶点被聚硅烷功能化的POSS为核心的支化单元,其结构如式2所示,其中“*”代表与式1连接之处。该聚合物是三维超支化共轭聚合物。
根据本发明的一个具体实施方式中,所述m为54,n为39。
而根据本发明的研究,优选的,本发明聚-(二卤甲基苯基硅烷-co-二卤二苯基硅烷)为无规聚合物。聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)的合成并不需要特别的限定,本领域的技术人员可以按照常规共聚硅烷的制备方法进行合成。
聚-(二卤甲基苯基硅烷-co-二卤二苯基硅烷)为聚-(二氯甲基苯基硅烷-co-二氯二苯基硅烷)或聚-(二溴甲基苯基硅烷-co-二溴二苯基硅烷)。
从共聚硅烷的溶解性及共聚硅烷对硝基芳烃具有富集作用来讲,优选的,所述聚-(二卤甲基苯基硅烷-co-二卤二苯基硅烷)为无规共聚硅烷。
经试验测定,上述超支化聚合物具有微孔和介孔结构,其中,介孔孔径分布在2~55nm之间,主要孔径分布在15~20nm(优选17nm)。该聚合物具有的丰富孔道结构增加了硝基芳烃的渗透性与接触面积,检测灵敏度更高。
本发明还提供上述硅基超支化共轭聚合物的制备方法,包括以下步骤:
将八(3-巯丙基)POSS和聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)通过巯基-烯点击化学反应合成所述的硅基超支化共轭聚合物。
八(3-巯丙基)POSS合成并不需要特别的限定,可以采用现有技术中的常规方法进行合成。但在本发明的优选实施方式中,所述八(3-巯丙基)POSS的合成方法,包括以下步骤:取浓HCl、去离子水、巯丙基三甲氧基硅烷,然后溶解在乙醇中,在N2环境中,于室温磁力搅拌水解,再转入油浴中磁力搅拌,使反应冷却,然后置于-8℃的冷冻器中过夜(8h~16h),得到白色沉淀,最后用乙醇洗涤,烘干,得到白色固体化合物I。其中:
进一步,所述浓HCl、去离子水、巯丙基三甲氧基硅烷的添加体积比例为1.0:5.0~9.0:3.0~5.0。进一步优选的体积比例为1.0:5.0:4.0。
进一步的,合成八(3-巯丙基)POSS的反应时间为3~4天,进一步优选的反应时间为3天。
聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)的合成并不需要特别的限定,本领域的技术人员可以按照常规共聚硅烷的制备方法进行合成。但在本发明的优选实施方式中,所述共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)的合成,包括以下步骤:利用伍尔兹反应(Wurtz反应),在钠沙甲苯分散体系中,将二卤甲基苯基硅烷和二卤二苯基硅烷共聚,回流;然后加入甲醇并收集粗产物,依次用甲苯,四氢呋喃(THF)和水洗涤,得到白色粉末状化合物II。其中:
为使得共聚硅烷与POSS产生有效协同作用,使得共聚硅烷的有效共轭长度增加,经过试验验证,进一步的,所述二卤甲基苯基硅烷和二卤二苯基硅烷的摩尔比例为1.0:0.5~1.5。进一步优选的摩尔比例为1.0:1.3。
进一步的,为了能够发生巯基-烯点击化学反应,共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)的基团末端需要添加官能团烯基,优选添加乙烯基,从而得到乙烯基共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)(化合物III)。乙烯基共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)的合成并不需要特别限定,本领域的技术人员根据所述化合物的结构和公知常识能够常规合成。但在本发明的优选实施方式中,所述乙烯基共聚(二卤甲基苯基硅烷)的合成方法,包括以下步骤:室温下,将乙烯基卤化镁(优选乙烯基氯化镁或乙烯基溴化镁)加入到共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)无水THF溶液中;并将溶液加热搅拌;通过蒸馏除去THF并浓缩产物;将剩余的反应混合物溶于二氯甲烷中,过滤除去副产物盐;最终,得到白色粉末状乙烯基共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)(即化合物III)。
进一步优选的,所述乙烯基卤化镁与共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)的添加比例为(0.1~1)mL:(0.1~0.15)g;最优选的加入量为0.1mL:0.13g。
进一步优选的,合成乙烯基聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)的反应温度为40~60℃,进一步优选的反应温度为60℃。
本发明中,所述巯基-烯点击化学反应中采用的反应溶剂为无水甲苯,采用此反应溶剂,更加有利于反应的进行。
本发明中,所述的巯基-烯点击化学反应中采用的自由基引发剂为偶氮二异丁腈
(AIBN),采用此引发剂,聚合效果优异。
本发明中,合成硅基超支化共轭聚合物的反应时间为4~8小时,进一步优选的反应时间为4小时;反应温度为60~80℃,进一步优选的反应温度为60℃。
本发明中,合成含σ共轭超支化聚合物的反应在惰性气体条件下进行,所述惰性气体可为氩气或氮气。
在本发明中,所述八(3-巯丙基)POSS、乙烯基共聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)和偶氮二异丁腈(AIBN)的质量比例为1.0:0.1~0.6:0.10~0.15,进一步优选的质量比例为1:0.6:0.10。
在本发明优选的一些实施方式中,具体包括以下步骤:
在惰性气体环境中,将八(3-巯丙基)POSS、AIBN和乙烯基聚(二卤甲基苯基硅烷-co-二卤二苯基硅烷)混合,油浴加热;然后除去油浴,将溶液暴露于空气中;降温,粗产物析出,用氯仿多次洗涤,合并滤液,在室温下静置8h~16h,干燥,回收滤液中产物,得到白色粉末状化合物(IV)。
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
实施例1
合成路线如图1所示:
(1)八(3-巯丙基)POSS(I)的制备方法:
在N2环境中,在三口烧瓶中加入乙醇(240mL),去离子水(17.5mL)和浓HCl(2.5mL,37%)搅拌、加热至回流。然后加入巯丙基三甲氧基硅烷(10mL)并继续回流1天。使反应冷却,然后置于-8℃的冷冻器中过夜,得到白色沉淀。过滤,回收白色沉淀,并将滤液在N2下收集到另一个烧瓶中。然后将白色沉淀物用乙醇洗涤几次,并在60℃的烘箱中干燥过夜(产率7%),然后将来自上面的滤液进一步回流3天,在8℃的冷冻箱中冷却过夜,产生更多的白色沉淀。如前所述过滤,滤液在90℃再回流24小时。使溶液冷却,然后置于-8℃的冷冻器中过夜,总产率为15%。得到白色固体(I)。
(2)共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(II)的合成:
无水甲苯(90mL)中加入金属钠(0.08mol,1.84g),并制成钠沙分散体。将二氯甲基苯基硅烷(0.04mol,9.55g)和二氯二苯基硅烷(0.05mol,12.70g)溶解在无水甲苯(90mL)中,然后将混合物缓慢滴加到钠沙分散体溶液中,回流12小时。然后加入少量甲醇并收集粗产物,依次用甲苯、THF和水洗涤。产率65%,产物为白色粉末(II)。
(3)乙烯基共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(III)的合成
将共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(PDMPS)(5×10
(4)三维超支化共轭聚合物(3D-HPs)(IV)的合成
在N2环境中,将八(3-巯丙基)POSS(0.050mol,52.80g)和AIBN(0.005mol,0.80g)溶于无水甲苯(0.25mL)中。然后加入乙烯基共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(0.006mol,60g),将烧瓶置于80℃的油浴中6小时。然后除去油浴,将溶液暴露于空气中。在约40℃下沉淀,回收产物。将反应烧瓶用氯仿洗涤,合并滤液在室温下静置过夜,将烧瓶在60℃下在真空烘箱中干燥4小时,得到白色粉末(IV)。
实施例2
(1)八(3-巯丙基)POSS(I)的合成:
在N2环境中,在三口烧瓶中加入乙醇(240mL),去离子水(12.5mL)和浓HCl(2.5mL,37%)搅拌、加热至回流。然后加入巯丙基三甲氧基硅烷(7.5mL)并继续回流1天。使反应冷却,然后置于-8℃的冷冻器中过夜,得到白色沉淀。抽滤,回收白色沉淀,并将滤液在N2下收集到另一个烧瓶中。然后将白色沉淀物用乙醇洗涤几次,并在60℃的烘箱中干燥过夜(产率3%),然后将来自上面的滤液进一步回流4天,随后在8℃的冷冻箱中冷却过夜,产生更多的白色沉淀。如前所述过滤,滤液在90℃再回流24小时。使溶液冷却,然后置于-8℃的冷冻器中过夜,总产率为11%,得到白色固体(I)。
(2)共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(II)的合成:
无水甲苯(90mL)中加入金属钠(0.08mol,1.84g),并制成钠沙分散体。将二氯甲基苯基硅烷(0.04mol,9.55g)和二氯二苯基硅烷(0.02mol,5.08g)溶解在无水甲苯(90mL)中,然后将混合物缓慢滴加到钠沙分散体溶液中,回流12小时。然后加入少量甲醇并收集粗产物,依次用甲苯,THF和水洗涤。产率50%,产物为白色粉末(II)。
(3)乙烯基共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(III)的合成:
将共聚(二氯甲基苯基硅烷)(PDMPS)(5×10
(4)三维超支化共轭聚合物(3D-HPs)(IV)的合成:
在N2环境中,将八(3-巯丙基)POSS(0.050mol,52.8g)和AIBN(0.003mol,0.48g)溶于无水甲苯(0.25mL)中。然后加入乙烯基共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(0.005mol,50g),将烧瓶置于60℃的油浴中6小时。然后除去油浴,将溶液暴露于空气中。通过在约40℃下沉淀回收产物。将反应烧瓶用氯仿洗涤,合并滤液在室温下静置过夜,将烧瓶在60℃下在真空烘箱中干燥6小时,得到白色粉末(IV)。
实施例3
(1)八(3-巯丙基)POSS(I)的合成:
在N2环境中,在三口烧瓶中加入乙醇(240mL),去离子水(22.5mL)和浓HCl(2.5mL,37%)搅拌、加热至回流。然后加入巯丙基三甲氧基硅烷(12.5mL)并继续回流1天。使反应冷却,然后置于-8℃的冷冻器中过夜,得到白色沉淀。通过在重力下过滤回收白色沉淀,并将滤液在N2下收集到另一个烧瓶中。然后将白色沉淀物用乙醇洗涤几次,并在60℃的烘箱中干燥过夜(产率5%),然后将来自上面的滤液进一步回流3.5天,随后在8℃的冷冻箱中冷却过夜,产生更多的白色沉淀。如前所述过滤,滤液在90℃下回流24小时。使溶液冷却,然后置于-8℃的冷冻器中过夜,总产率为13%。得到白色固体(I)。
(2)共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(II)的合成:
无水甲苯(90mL)中加入金属钠(0.08mol,1.84g),并制成钠沙分散体。将二氯甲基苯基硅烷(0.04mol,9.55g)和二氯二苯基硅烷(0.06mol,19.05g)溶解在无水甲苯(90mL)中,然后将混合物缓慢滴加到钠沙分散体溶液中,回流12小时。然后加入少量甲醇并收集粗产物,依次用甲苯,THF和水洗涤。产率52%,产物为白色粉末(II)。
(3)乙烯基聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(III)的合成:
将共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(PDMPS)(5×10
(4)三维超支化共轭聚合物(3D-HPs)(IV)的合成:
在N2环境中,将八(3-巯丙基)POSS(0.050mol,52.8g)和AIBN(0.002mol,0.32g)溶于无水甲苯(0.25mL)中。然后加入乙烯基共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(0.007mol,75g),将烧瓶置于70℃的油浴中6小时。然后除去油浴,将溶液暴露于空气中。在约40℃下沉淀,回收产物。将反应烧瓶用氯仿洗涤,合并滤液在室温下静置过夜,将烧瓶在60℃下在真空烘箱中干燥8小时,得到白色粉末(IV)。
实施例4实施例1中的聚合物的基本性质
(1)八(3-巯丙基)POSS(I)、共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(II)和三维超支化共轭聚合物(3D-HPs)(IV)的理化性质
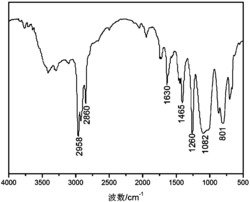
八(3-巯丙基)POSS(I)、共聚(二氯甲基苯基硅烷-co-二氯二苯基硅烷)(II)和三维超支化共轭聚合物(3D-HPs)(IV)的红外波谱对比图如图2。可以看出,三维超支化共轭聚合物(3D-HPs)(IV)的红外光谱中在1122and 1025cm
化合物IV的GPC测定结果显示:数均分子量为4.107×10
化合物IV热稳定性测试显示在341℃时开始失重,较化合物II的热分解温度(271℃)高,一方面说明引入具有较高热稳定性的POSS结构可以增大其热稳定性;另一方面,说明化合物IV具有较好的热稳定性,且在低沸点的四氢呋喃中有很好的溶解性,有利于成膜。
将化合物IV研磨成细小粉末,对化合物IV进行X射线衍射。结果如图3所示,分析其衍射图谱,在2θ≈20°处出现明显的峰型,说明化合物具有T8-POSS笼型结构。根据布拉格方程(2dsinθ=nλ)计算得,晶面间的距离d为1.5nm,这一实验结果证实了化合物IV是具有笼型结构的三维分子。
(2)氮气吸附等温线的测试
化合物IV的环境扫描电镜图如图4所示。
取100mg化合物IV在150℃下脱气至少24小时,用UHP级氮气(99.999%)气源于77K温度下,对化合物进行氮气吸附的测量,结果如图5所示。分析其氮气吸附等温线,回滞环属于II型,说明该化合物IV为微孔和介孔结构。从图5B可以看出,孔径分布较宽,主要孔径为17nm。这一实验结果说明化合物IV是具有微孔和介孔结构的三维孔道结构分子。
(3)化合物IV和化合物II的吸收和发射性能
如图6所示,化合物IV和化合物II的紫外吸收(A)和荧光发射光谱(B)。从图6(A)可以看出,化合物IV的最大吸收峰比化合物II的最大吸收峰红移了接近14nm,这是由于化合物IV与化合物II相比,共轭体系增加,降低了最高占据分子轨道(HOMO)与最低未占分子轨道(LUMO)之间的能级差。从吸收谱图中可以看出,化合物IV分子间没有发生明显的π-π堆积。从图6(B)的荧光光谱图中可以看出,化合物IV的荧光峰与化合物II相比也发生了显著的蓝移,且化合物IV的stokes位移达到75nm,这是由于超支化共轭聚合物具有多维树突结构,侧链的旋转与振动消耗激发态能量导致,即在受到光子激发后,发生了较大程度的共振弛豫,引起荧光能量下降。较大的stokes位移是荧光传感材料的必要条件,因此三维超支化共轭聚合物(3D-HPs)比一维线型聚合物(II)相比具有更大的优势。
实施例5化合物IV便携式试纸对溶液中的痕量TNT的检测应用
测试方法
1)溶液配制:称取10mg化合物IV溶于100ml THF中,配制1×10
2)以化合物IV制备的便携式试纸对溶液中的痕量TNT的检测步骤:
用玻璃点样毛细管(0.3mm)分别吸取上述(7、14、21、28、35、42、49、56、63和70μM)TNT溶液在化合物IV便携式试纸的中心点样,过程操作均在三用紫外分析仪(365nm)照射下进行,结果如图11(A)所示。
可以看出,对比空白的化合物IV便携式试纸,随着TNT溶液浓度的增加,试纸中心被猝灭的黑点越来越深,值得注意的是,痕量TNT溶液(7μM)时,中心被猝灭黑点就能用裸眼看到,说明该试纸能灵敏地检测痕量TNT溶液。如图11(B)所示,在TNT溶液70μM时,化合物IV溶液已被完全猝灭。这不仅增加了荧光共轭聚合物检测痕量硝基芳烃溶液的实用性,而且具有方法简单、材料易得等特点。
实施例6荧光传感器的制备
薄膜通过Spin Coater KW-4A型涂布机在玻璃基质(10×20×1mm
化合物IV和化合物II的溶液的浓度均为0.1g/L,转速为2000rpm。待涂布机匀速以后取50uL溶液逐滴滴加到玻璃片上,使用前将薄膜在空气中自然晾干,所述薄膜即为荧光薄膜传感器的传感材料。
以下实施例中所用的化合物为实施例1中制备得到的。
实施例7荧光薄膜传感器的光学性质
分别测化合物II、IV旋涂薄膜的紫外可见吸收和荧光发射光谱,结果如图6所示。可以看出,无论对于化合物II还是IV,其旋涂薄膜的吸收峰较溶液中相应的化合物范围宽且红移,说明薄膜中有一定的堆积作用。然而引入三维POSS结构的化合物IV旋涂膜与化合物II旋涂薄膜相比,其吸收光谱发生蓝移,说明具有空间效应的POSS结构能减少一部分堆积作用。化合物II和IV在溶液和薄膜状态的荧光发射图谱中也表现出相似的规律。
实施例8化合物荧光传感性能的测试
(1)在THF溶液中化合物II对TNT的荧光猝灭性能
测试方法
1)溶液配制:称取10mg化合物II溶于100ml THF中,配制1×10
2)化合物II对TNT传感性能测试步骤:依次加入不同浓度的TNT溶液,并测试其荧光光谱,如图7(A)所示。并记录荧光峰强度,计算化合物II在不同浓度TNT溶液中的猝灭效率(ηEP),结果如图8所示。
(2)在THF溶液中化合物IV对TNT的荧光猝灭
测试方法
1)溶液配制:称取10mg化合物IV溶于100ml THF中,配制1×10
2)化合物IV对TNT传感性能测试步骤:将上述配置的化合物IV的THF溶液3ml(浓度为1×10
可以看出,随着TNT浓度的升高,化合物IV溶液的荧光强度逐渐减弱。当TNT浓度为1.02×10
Stern-Volmer猝灭常数(KSV)是评价传感器材料灵敏度的重要指标,如图8所示。对比化合物II与化合物IV的KSV发现,引入POSS结构的化合物IV(KSV=2.83×10
(3)化合物II和化合物IV薄膜传感器在饱和DNT蒸汽中的荧光淬灭
化合物II旋涂膜传感器对气相DNT的传感性能测试:
测试方法:
将化合物II旋涂薄膜沿荧光池对角线小心插入(玻璃片与入射光的夹角为45°)。测定旋涂薄膜在空气中的荧光强度随时间的变化情况(λex/λem=347/425nm)。将薄膜取出,并向荧光池中加入100mg DNT粉末,加盖密封后,室温下静置l h,使荧光池内DNT蒸汽达到饱和。然后将旋涂薄膜沿荧光池对角线小心插入(玻璃片与入射光的夹角为45°),并测定旋涂薄膜在含有DNT的空气中的荧光强度随时间的变化情况(λex/λem=347/425nm)。测试结果如图9(A)所示。
如图10所示,旋涂薄膜一旦插入DNT饱和蒸汽中,其荧光强度瞬间降低到其在空气中荧光强度的30%(猝灭效率70%),说明该单分子膜传感器荧光强度对DNT蒸汽具有响应性,但猝灭效果不佳。
化合物IV旋涂薄膜对气相DNT的传感性能测试:
测试方法:将化合物IV旋涂薄膜沿荧光池对角线小心插入(薄膜与入射光的夹角为45°)。测定旋涂薄膜在空气中的荧光强度随时间的变化情况(λex/λem=330/405nm)。将薄膜取出,并向荧光池中加入100mg DNT粉末,加盖密封后,室温下静置l h,使荧光池内DNT蒸汽达到饱和。然后将旋涂薄膜沿荧光池对角线小心插入(薄膜与入射光的夹角为45°),并测定旋涂薄膜在含有DNT的空气中的荧光强度随时间的变化情况(λex/λem=330/405nm)。测试结果如图9(B)所示。
如图10所示,旋涂薄膜一旦插入DNT饱和蒸汽中,其荧光强度瞬间降低到其在空气中荧光强度的18%(猝灭效率82%),说明该单分子膜传感器荧光强度对DNT蒸汽响应灵敏,可迅速达到其猝灭响应平衡,且300s内,基本可以完全猝灭。而目前常见的薄膜传感器往往需要一定的时间才能达到猝灭响应平衡,猝灭效率低。引入POSS结构,三维孔道结构,增加了DNT分子的渗透性,使得猝灭剂分子与传感单元充分接触。尤其是聚硅烷上,带正电性的硅原子能与硝基芳烃N/O原子形成非共价键,对硝基芳烃具有富集作用。可见引入POSS结构的化合物IV旋涂膜玻璃片传感器在响应速度方面表现出了显著的优势。
(4)化合物IV薄膜的循环性测试
在饱和DNT蒸汽压环境下,聚合物薄膜的循环性测试方法如下:先记录没有猝灭的聚合物薄膜的荧光光谱,然后将薄膜传感器放入达到DNT饱和蒸汽压的比色皿中检测其荧光光谱并记录;接着将薄膜传感器用N2吹扫多次,再次记录薄膜传感器的荧光光谱。如此循环重复5次。荧光光谱峰值得变化如图12所示。在图中可以发现,第1次循环后聚合物薄膜的荧光强度下降了12.7%,而第5次循环后聚合物薄膜的荧光强度下降了19.6%。从第一次到第五次循环,猝灭效率从81.99%降到77.5%。从测试结果来看该聚合物薄膜传感器具有较好的循环性。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
一种硅基超支化共轭聚合物及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0