









专利摘要
本发明是改进的循环的、吸热的氢转化工艺以及用于实现该工艺的催化剂床系统。具体地,改进工艺包括使烃与多组分催化剂床以这样的方式反应,该方式使得催化剂床内的温度在整个工艺的所有阶段中保持在受控温度范围内。多组分催化剂床包括与产热材料物理混合的反应特异性催化剂。
说明书
技术领域技术领域
本发明涉及改进的循环的、吸热的烃转化工艺以及涉及用于实现该工艺的催化剂床系统。具体地,改进工艺包括使烃原料与多组分催化剂床接触,其中催化剂床包括第一组分和第二组分,所述第一组分为被特定地设计为将烃进料转化为预定产物或产物混合物的催化剂,所述第二组分在被暴露于还原反应条件和/或氧化反应条件之后产生热。
技术背景背景技术
多种吸热烃转化工艺被用于商业操作。这些工艺包括Houdry循环固定床脱氢工艺、流化床链烷烃脱氢工艺、流化床乙苯脱氢工艺和流化床催化裂化工艺、以及其他工艺。由于这些工艺是吸热的,必须从周围消耗热以便发生烃转化反应。在这些工艺的每一种中,至少一种反应是通过使烃进料与催化剂接触来促进的。此外,在这些工艺的每一种中,存在再生催化剂的至少一种还原反应和/或氧化反应。发生吸热反应所需要的热部分地通过在转化工艺期间沉积在催化剂上的焦炭和其他不期望的副反应物的燃烧来提供。这一燃烧在再生工艺期间发生。然而,通常需要另外的热,且这由在烃转化循环之间从外部源被供给到催化剂床的热空气或蒸汽提供。
作为例子,在美国专利2,419,997中教导的典型Houdry脱氢工艺中,脂族烃经过脱氢催化剂床。当脂族烃通过催化剂床时,该烃被脱氢成其互补烯烃。然后,从催化剂床除掉该烯烃,该催化剂被再生且被还原,且重复此循环。这一脱氢反应是高度吸热的。因此,在脱氢步骤期间,接近于催化剂床入口(脂族烃最初进入催化剂床的地方)的温度可被降低多达100℃。这一温度降低引起烃转化的降低。此外,在脱氢步骤期间,焦炭通常形成并沉积在催化剂上,这进一步降低了催化剂的活性。
为了再加热催化剂床并移除已经沉积在催化剂上的焦炭,使用被加热到高达700℃的温度的空气,反应器被清除烃,且然后经历再生步骤。由通过该床的热空气且也由沉积在催化剂上的焦炭的燃烧来向该床提供热。使用诸如氢气的还原气体,在脱氢步骤之前的催化剂的还原也提供一些热。在再生期间,热空气从催化剂床的入口流到出口。这一再生循环通常相对短,因此存在该床的入口显著地比该床的出口热的倾向,但由于Houdry脱氢工艺中的循环之间的定时,催化剂床并没有时间来进行热平衡。因而,当脂族烃被再次供给到反应器中时,该床的出口区域仍比该床的入口区域冷。该床的入口的高温易于引起不期望的副产物的形成,且因而降低了所期望的烯烃的选择性和收率。另一方面,该床的出口的较低温度并不允许催化剂的完全利用,且因而烯烃收率低于另外预期或期望的收率。而且,由于催化剂床中的焦炭分布并不是独立可控参数,所以该床内的热分布也不容易控制。这些因素中的每一种影响所得到的催化剂床温度分布并使得难以控制该床内的温度分布。
在美国专利2,423,835中,Houdry教导,通过在催化剂床内包括能够吸收或储存热的、可以随后按照期望或需要被释放的“惰性”物质,催化剂床温度可以被控制在适于进行反应的温度范围内,而不要求外部加热流或冷却流循环通过或环绕反应室。在用于固定床反应器的商业实践中,这通常通过把脱氢催化剂和粒状α-氧化铝“惰性”物质的物理混合物用作催化剂床来获得。尽管惰性物质的添加为该工艺提供可逆的吸热装置,并帮助稳定反应器中的全局温度振荡,但该惰性物质并既不能为该工艺提供额外的热,也不能在该工艺的任何阶段期间产生热。因此,即使有催化剂和惰性物质的组合使用,仍然需要额外的热源。
难题在于识别用于控制吸热工艺的催化剂床中的温度分布的商业可行方法。理想地,任何此类方法将允许把热增加到催化剂床的预定部分而不需要使用产生大量不需要的副产物的催化活性材料。
发明内容发明内容
本发明是改进的吸热的烃转化工艺以及用于实现该工艺的催化剂床系统。具体地,改进工艺包括使烃与多组分催化剂床以这样的方式反应,该方式使得催化剂床内的温度在整个工艺的所有阶段中保持在受控温度范围内。多组分催化剂床包括与产热材料物理混合的反应特异性催化剂。任选地,可以将如本领域已知的惰性物质与催化剂和产热材料进一步物理地组合。产热材料以这样的方式将热添加到催化剂床,该方式使得出口区域的床被保持在足够高的温度以有效地将烃转化为烯烃。在一个示例性的实施方案中,所述工艺为Houdry脱氢工艺,反应特异性催化剂为常规铬基脱氢催化剂,且产热材料为载于铝酸钙载体上的氧化铜,且任选地存在的惰性物质为α-氧化铝。
工业实用性
本文描述的本发明可以用于涉及吸热烃转化的任何操作。这些工艺包括例如Houdry循环固定床脱氢工艺、流化床链烷烃脱氢工艺、流化床乙苯脱氢工艺和流化床催化裂化工艺、以及其他工艺。
附图说明附图说明
图1为在将丙烷转化为丙烯期间装载有55vol.%(体积百分比) 标准催化剂和45vol.%α-氧化铝的绝热反应器中温度分布的图示。
图2为在将丙烷转化为丙烯期间绝热反应器中温度分布的图示,其中反应器被装在分为约35%出口、30%中间和35%入口的三个区域,且其中床的出口区域和入口区域包含55vol.% 标准催化剂和45vol.%α-氧化铝,且床的中间区域包含55vol.% 标准催化剂和铝酸钙载体上的45vol.%氧化铜。
具体实施方式具体实施方式
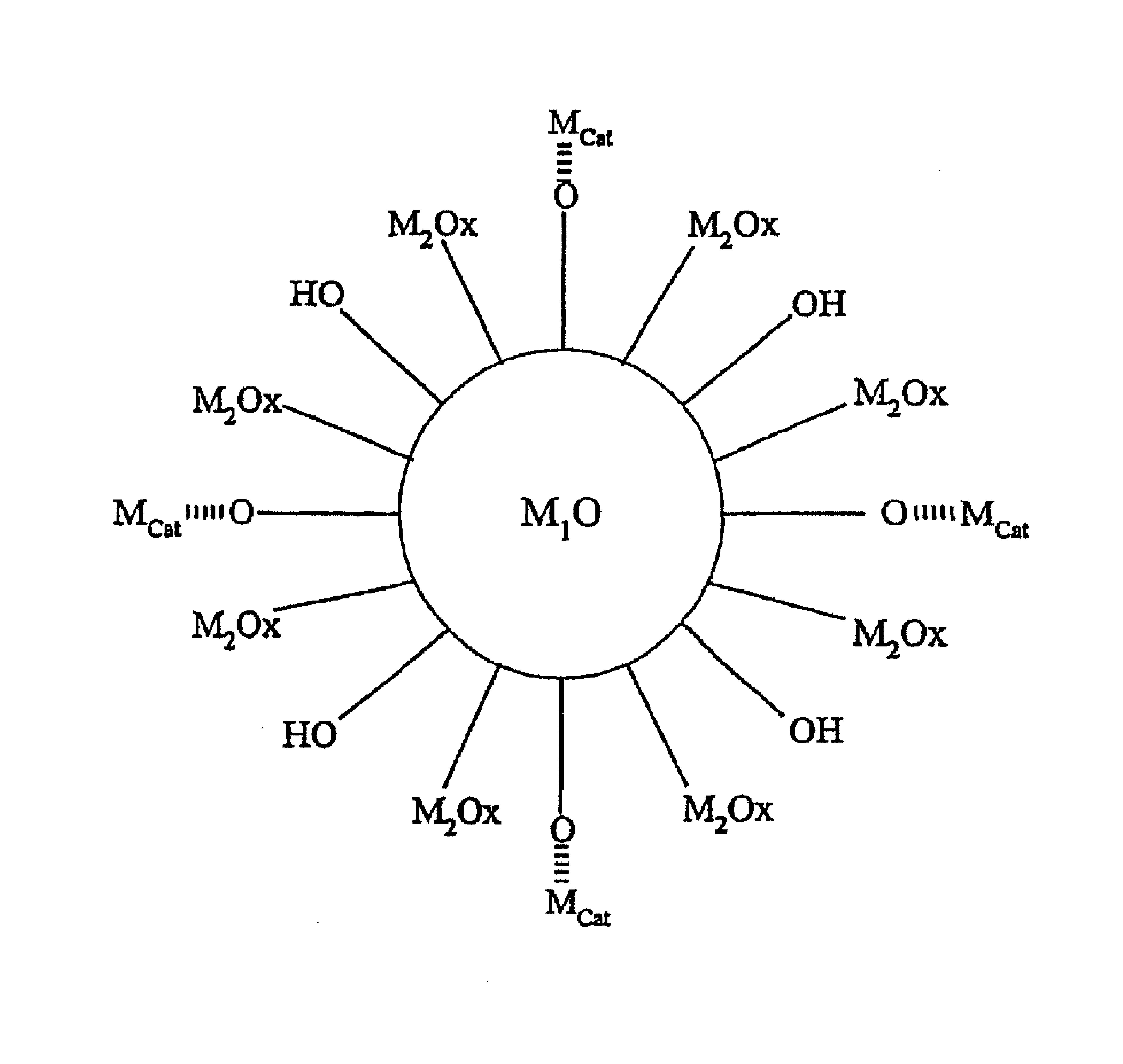
本发明的改进工艺期望用于任何循环的吸热的烃转化工艺,无论是在固定床还是在流化床应用中。改进工艺要求烃原料接触多组分催化剂床以便实现吸热烃转化,并要求然后催化剂床暴露于氧化和/或还原条件。催化剂床包括催化第一组分和产热第二组分。催化第一组分必须是被特定地设计为经由吸热反应将烃进料转化为预定产物或产物混合物的催化剂。产热第二组分必须是在被暴露于还原和/或氧化反应条件之后产生热但关于不期望的副反应如焦炭形成相对于烃原料来说惰性的材料。任选地,产热组分可以将烃催化转化为所需的产物或产物混合物。多组分催化剂床还可任选地包括用于催化剂床的惰性物质,如本领域中已知的。
为了详细描述本发明的目的,使用固定床反应器的Houdry循环脱氢工艺的改进将用作实施方案。然而,应理解,在没有偏离本发明的范围的情况下可以修改本发明,以便以发明方式在其他循环的吸热烃转化工艺中起作用,其他循环的吸热烃转化工艺例如,但不限于,流化床链烷烃脱氢工艺和流化床乙苯脱氢工艺。
用于Houdry循环脱氢工艺的装置包括含有固定催化剂床的绝热反应器,其中床界定了入口区域和出口区域。催化剂床包括催化第一组分和产热第二组分。任选地,如本领域已知的,还可将惰性物质添加到催化剂床。
为了描述本发明的目的,但不期望通过这样的描述进行任何限制,固定催化剂床基本上被分成三个大约相等的部分-床的入口区域、床的中间区域、床的出口区域。除非另有说明,对于本文呈现的任何实例来说,与惰性物质组合的催化第一组分用于床的入口区域和出口区域;且与产热第二组分组合并与或不与惰性物质一起使用的催化第一组分用于床的中间区域。尽管便于描述的目的,但应理解,组分组合的若干变化对于固定催化剂床来说是可能的。例如,当床被分成三个区域时,床可以被分成,使得(a)三个区域中的每一个具有大约相等的体积,或者(b)中间区域可以大于总催化剂体积的约三分之一,或者(c)中间区域可以小于总催化剂体积的约三分之一,或者(d)入口区域和出口区域可以具有不相等的体积,或者(e)其任何组合。此外,并不必须将床分成三个区域。例如,脱氢催化剂可以与产热材料混合并与或不与另外的惰性物质一起使用,且然后混合物可以被装入到没有分区的反应器中。可选择地,可以将催化剂床分成两个区域,且分别地,与惰性物质组合的脱氢催化剂在入口区域或在出口区域,而与产热材料组合并与或不与另外的惰性物质一起使用的脱氢催化剂在出口区域或入口区域。在一些情况下,将催化剂床分成多于三个区域还可能是有益的,且任选地与惰性物质组合的脱氢催化剂层交替与或不与另外的惰性物质一起使用的、与产热材料组合的脱氢催化剂层。
然而,如本领域已知的,对于催化剂床取向具有一些实际限制。例如,已知如果催化剂床的区域变得太热,那么存在反应失控的危险。因此,使用者将被很好地建议知道在没有添加产热材料的情况下的感兴趣的反应的温度分布,并将这用作指导以确定产热材料在催化剂床内何处将是最有效的。此外,在任何特定的区被添加到催化剂床的产热材料的量将由在整个催化剂床的工艺中必须被替代的热的量确定。换句话说,由产热材料产生的热必须小于在催化剂床的每一个部分中由主要反应消耗的热。不正确地将产热材料集中在床的一个区域中会导致显示比没有使用产热材料的工艺的温度分布更大温度偏差的温度分布。
示例性的工艺通常遵循如美国专利2,419,997中描述的典型Houdry脱氢工艺。Houdry工艺包括一系列阶段,其中催化剂床被抽空、用氢气还原并被抽空,然后脂族烃被引入并脱氢,然后催化剂床被蒸汽吹扫并再生,且循环从还原阶段开始重复。
在发明工艺中,催化剂床被抽空并用诸如氢气的还原气体还原。在该阶段期间,反应器床中的产热第二组分产生进入反应器床的催化第一组分中的另外的热。然后,脂族烃被供入到催化剂床中并在与反应器床的催化第一组分接触时被脱氢。因为床的催化第一组分已经基本上被产热第二组分预热,所以催化第一组分证明了相对于不包括产热第二组分的反应器床的改进的转化。然后,催化剂床被蒸汽吹扫并再生,且循环从还原阶段开始重复。在再生步骤期间,产热第二组分还可以产生另外的热。在一个优选的实施方案中,产热第二组分被选择为使得没有观察到对烃转化反应的选择性具有明显的负影响。
在发明的脱氢工艺中,催化第一组分可以是被设计为用于脱氢反应的任何催化剂,如可从Süd-Chemie Inc.,Louisville,KY得到的 标准催化剂。 标准催化剂为在氧化铝载体上制造的氧化铬脱氢催化剂,其包括约17wt%(重量百分比)到约22wt%Cr2O3。
产热第二组分必须是在被暴露于还原和/或氧化反应条件之后产生热但关于烃转化为不期望产物或关于不期望的副反应相对惰性的材料。产热第二组分包括选自由以下组成的组的金属:铜、铬、钼、钒、铈、钇、钪、钨、锰、铁、钴、镍、银、铋及其组合。产热第二组分的示例性载体包括但不限于,各种铝的氧化物或氢氧化物,如三氢氧化铝、勃姆石、假勃姆石、三水铝石、三羟铝石、过渡型氧化铝或α-氧化铝、二氧化硅/氧化铝、二氧化硅、硅酸盐、铝酸盐如铝酸钙或六铝酸钡、煅烧水滑石、沸石、氧化锌、氧化铬、氧化镁及其组合。任选地,产热第二组分还可以包括促进剂,例如碱、碱土金属、锂、钠、钾、铷、铯、铍、镁、钙、锶、锆、钡及其组合。
金属构成产热第二组分总重量的约1wt%到约100wt%。在一个更优选的实施方案中,金属构成第二组分总重量的约2wt%到约40wt%;且在一个最优选的实施方案中,金属的量为第二组分总重量的约5wt%到约10wt%。
产热第二组分通过本领域已知的用于制备载体催化剂的基本上相同的方法来制备。例如,产热第二组分可以通过用金属沉淀载体或通过用金属浸渍第二载体来制备。促进剂还可以与金属一起添加,或可以经由本领域已知的用于添加促进剂的方法以其他方式添加到第二组分中。
在装入反应器中之前,催化第一组分可以与惰性物质物理混合,如本领域已知的。该惰性物质可以是任何这样的物质或物质的组合,即它们关于不期望的副反应在催化上是无活性的,且具有高密度和高热容,但在工艺的任何阶段期间不能产生热。通常使用的惰性物质为具有与被载催化第一组分相似粒度的粒状α-氧化铝物质。此外,如本领域已知的,惰性物质和催化第一组分之间的体积比取决于多种因素,所述因素包括但不限于,用于脱氢工艺中的烃进料的类型。在本应用中,没有规定特定的体积比,而是使用者可以将该比调节为适合于期望用途。
对于固定床应用,催化剂床通过将催化第一组分和/或惰性物质和/或产热第二组分物理混合来制备。最初,界定催化第一组分的期望量和期望的床构型。然后,将催化第一组分分成界定量并与产热第二组分或与惰性物质或与产热第二组分和惰性物质的组合物理混合。然后,将混合物装入反应器的每一期望床构型。产热第二组分不会影响所添加的催化剂的量,也不会影响所得到的催化剂床中的催化剂与惰性物质的相对比。
本开发的催化剂床系统还可以用于移动流化床操作。在仅仅为了示例而呈现且不期望是限制性的一个示例性流化床操作中,脱氢处理系统具有两个平行的绝热反应器-用于脱氢反应的第一反应器和用于再生的第二反应器。系统通过如下方式来操作:将脱氢催化剂装到第一反应器中,在第一反应器中提供约15分钟的停留时间,然后将装载的催化剂移动到第二反应器中并提供约15分钟的停留时间,且然后将催化剂返回到第一反应器中并继续交替反应器工艺。在再生反应器中,将催化剂在650℃的空气下处理并由脱氢步骤期间形成的燃烧焦炭的热和由热空气再加热。当将催化剂从第二反应器移回到第一反应器中时,使催化剂经受还原环境以制备用于另一个脱氢反应的催化剂。
在移动流化床操作中,脱氢催化剂通常不与惰性物质混合,且当工艺循环通过各个阶段时,观察到相对宽的温度摆动。然而,使用发明方法,将脱氢催化剂与产热第二材料组合并均匀地装入到脱氢反应器中。然后将催化剂加上产热材料移动到再生反应器中,在再生反应器中焦炭烧尽反应特异性催化剂。当催化剂加上产热材料返回到脱氢反应器中时,产热材料通过还原环境来活化并将足够的热加入到催化剂床,以相比于在不包括产热材料的流化床催化剂负载中的转化率水平,增加烃转化的量。
以下为用于在流化床和固定床应用中制备和使用催化第一组分和产热第二组分的本发明的代表性实施例。这些实施例被呈现为进一步解释本发明,并不被期望或被用来限制本发明的范围。
材料制备
实施例A:将以商品名 标准催化剂销售并从Süd-Chemie Inc.,Louisville,KY可得到的催化第一组分用于商用脱氢单元180天。催化剂具有基于催化剂总重量的约19wt%氧化铬浓度。
实施例B:制备了具有约75μm平均粒度的氧化铬/氧化钾/γ-氧化铝脱氢催化剂。催化剂具有基于催化剂总重量的17.5wt%的氧化铬浓度和1.0wt%氧化钾浓度。
实施例C:产热第二组分根据本发明如下来制备:将α-氧化铝载体用饱和硝酸铜溶液浸渍,然后将浸渍过的载体在120℃下干燥,随后在空气-蒸汽气氛中在750℃下煅烧。产热第二组分具有基于第二组分的重量的约11wt%的CuO浓度。
实施例D:产热第二组分根据本发明如下来制备:将铝酸钙(Ca-铝酸盐)制粒为约3.5mm小丸,然后将Ca-铝酸盐在约1300℃下煅烧约10小时,然后将煅烧过的材料用饱和的硝酸铜和硝酸锰溶液浸渍,且将浸渍的材料在约250℃下干燥约4小时,随后在约500℃下煅烧约5小时。产热第二组分具有基于第二组分的重量的约11wt%的CuO浓度和约0.5wt%的MnO2浓度。
实施例E:产热第二组分根据本发明如下来制备:将勃姆石氧化铝与氧化钙混合,并将混合物制球以制备6mm直径的小丸,将该小丸在120℃下干燥并然后在1300℃下煅烧,该小丸具有18wt%的最终CaO含量。煅烧过的制粒材料用饱和的硝酸铜和硝酸锰溶液浸渍,且将浸渍的材料在约250℃下干燥,随后在1400℃的空气下煅烧。产热第二组分具有基于第二组分的重量的约11wt%的CuO浓度和约0.5wt%的MnO2浓度。
实施例F:产热第二组分根据本发明如下来制备:将三水合氧化铝(三水铝石)制粒为约5mm小丸,然后将三水铝石在约550℃下煅烧约4小时,然后将煅烧过的材料用饱和硝酸铜溶液浸渍,并将浸渍的材料在约250℃下干燥约4小时,随后在约500℃到1400℃下煅烧。产热第二组分具有基于第二组分的重量的约11wt%的CuO浓度和约0.5wt%的MnO2浓度。
实施例G:产热第二组分根据本发明如下来制备:将具有约75μm平均粒度的γ-氧化铝载体用饱和的硝酸铜和硝酸锰溶液浸渍,且然后将浸渍的材料在约250℃下干燥,随后在750℃的空气下煅烧。产热第二组分具有基于第二组分的重量的约8wt%的CuO浓度和约0.4wt%的MnO2浓度。
实施例H:将实施例A的催化第一组分与惰性α-氧化铝以55vol.%第一组分/45vol.%α-氧化铝的比物理混合。
实施例I:将实施例A的催化第一组分与实施例E的产热第二组分以55vol.%第一组分/45vol.%产热组分的比物理混合。
实施例J:将实施例B的催化第一组分与实施例G的产热组分以80vol.%第一组分/20vol.%产热组分的比物理混合。
实施例K:将新鲜的 标准催化剂的催化第一组分与实施例F的产热第二组分以55vol.%第一组分/45vol.%产热组分的比物理混合。
实施例L:将新鲜的 标准催化剂的催化第一组分用饱和硝酸铜溶液浸渍,且将铜浸渍的铬基催化剂在120℃下干燥并在空气-蒸汽气氛中在750℃下煅烧。铜浸渍的催化剂具有基于催化剂总重量的17.5wt%的氧化铬浓度和11wt%的氧化铜浓度。
实施例M:现有技术催化剂根据WO 02/068,119的实施例1来制备。通过将860g勃姆石氧化铝、800g氢氧化铜碳酸盐、120g乙酸钡、100gCrO3、700g NH4HCO3和250g去离子水组合在Eirich混合器中来制备催化剂。形成直径约3mm的颗粒并将其在120℃下干燥8小时以及在650℃的炉中煅烧10小时。铜浸渍的催化剂具有基于催化剂总重量的45wt%的氧化铬浓度和40wt%的氧化铜浓度。
实施例N:现有技术催化剂根据美国专利5,108,973的实施例1来制备。通过将763.8g氧化铝溶胶(包含7.51%Al2O3)和89.3g六水合硝酸铬共混在一加仑的搅拌机中,直到固体溶解,来制备催化剂。将六水合硝酸铜(116.3g)溶解在200ml去离子水中并将其加入到搅拌机中。然后,将61.8mol硼酸溶解在350ml温和的去离子水中并将其也加入到搅拌机中。将混合物共混另外的两分钟,直到混合物变成均匀的和深蓝色。然后,将700ml于甲醇溶液中的20%氢氧化铵加入以形成稠凝胶。将凝胶放在塑料盘上干燥,并在180℃下干燥4小时,且然后通过以下顺序来煅烧:25℃下2小时,175℃下12小时,400℃下4小时,830℃下8小时,830℃下4小时,250℃下3小时且然后冷却到室温。将煅烧过的材料制片以形成3mm直径的颗粒。铜浸渍的催化剂具有基于催化剂总重量的19wt%的氧化铬浓度和25wt%的氧化铜浓度。
性能测试
实施例1和实施例2:测试催化剂组合对于在具有约3600cc的催化剂床体积的下行绝热反应器中的丙烷到丙烯的转化率。将丙烷和空气通过入口供入到反应器中,且从出口回收丙烯。在1.0的液时空速下进行工艺,且丙烷温度为540℃到600℃,且空气温度为540℃到620℃,且空气与烃的比为7.1wt/wt。反应器以对于Houdry工艺常用的循环模式操作,且循环时间为60秒用于由氢气还原,540秒用于脱氢,60秒用于抽空,540秒用于再生-再热-氧化以及60秒用于抽空。反应器在循环的脱氢步骤期间在0.5atm的压力下操作,且在循环的再生步骤期间在大气压下操作。将循环操作重复300次。
实施例1:反应器加载-100vol.%实施例H的催化剂组合。
实施例2:反应器加载-将约35vol.%来自实施例H的材料在下行绝热反应器的出口附近加载,然后将约30vol.%来自实施例I的材料加载到反应器中间区域,然后将约35vol.%来自实施例H的材料在入口附近加载。
表1:丙烷脱氢中的催化剂的性能特征(绝热固定床反应器)
实施例3和实施例4:测试催化剂组合对于在具有约75cc的催化剂床体积的假绝热流化床反应器中的异丁烷到异丁烯的转化率。将异丁烷和空气通过入口供入到反应器中,且从出口回收异丁烯。在-3.34的液时空速下进行工艺,且异丁烷和空气温度为550℃到590℃,且空气与烃的比为3.5wt/wt。反应器以循环模式操作,且循环时间为60秒用于由氢气还原,540秒用于脱氢,60秒用于氮气吹扫,540秒用于氧化以及60秒用于氮气吹扫。反应器在循环的脱氢步骤和再生步骤期间在大气压下操作。将循环操作重复30次。
实施例3:反应器加载-100vol.%实施例B的催化剂组合。
实施例4:反应器加载-100vol.%实施例J的催化剂组合。
表2:异丁烷脱氢中的催化剂的性能特征(假绝热流化床反应器)
实施例5-实施例8:在具有约30cc的催化剂床体积的等温固定床反应器中测试催化剂组合对异丁烷到异丁烯的转化率。将异丁烷和空气通过入口供给到反应器中,且从出口回收异丁烯。在537℃、567℃和593℃的温度且在-2/hr的液时空速(LHSV)进行脱氢反应。
实施例5:反应器加载-100vol.%实施例K的催化剂组合。
实施例6:反应器加载-100vol.%实施例L的催化剂组合。
实施例7:反应器加载-100vol.%实施例M的催化剂组合。
实施例8:反应器加载-100vol.%实施例N的催化剂组合。
表3:异丁烷脱氢中的催化剂的性能特征(等温固定床反应器)
图1和图2分别显示实施例1和实施例2的催化剂床的温度分布。如由这些图所证明的,当在Houdry脱氢工艺期间产热第二组分被包括在固定催化剂床时,贯穿整个催化剂床中的催化剂床温度更为一致。在无产热第二组分的情况下,入口区域的温度的起伏覆盖约75℃的范围,而出口区域的温度的起伏覆盖仅约5℃的范围。此外,该床的出口区域的温度保持在约560℃-低于具有来自催化剂的最优转化性能所期望的温度。在有产热第二组分的情况下,该床的入口区域和出口区域两者在循环工艺期间都经历约45℃的温度起伏,但入口区域的平均温度为约580℃而出口区域的平均温度为约625℃,这提供了更高的催化剂总效率。如表1中所显示的,这转变成显著更高的转化而不牺牲选择性。
类似地,如表2中所显示的,当产热第二组分被用于流化床系统时,也看到转化率的改善。尽管流化床应用中的转化率的增加并不像在固定床应用中那样显著,但除了转化的增加之外,流化床应用并不显示出选择性的直接增加,这表明总的工艺比并不包括产热组分的现有技术的催化剂床更为有效。
令人惊奇的是,如表3的结果所显示的,当在脱氢催化剂组合物中组合了铜和铬时(实施例6),来自等温单元中脱氢工艺的转化和收率显著低于当铜作为与氧化铬脱氢催化剂分离但与其物理混合的组分出现在催化剂床时的情形(实施例5)。使用更高浓度的氧化铬和/或氧化铜(实施例7和实施例8)并不改变这些总的发现。
可以预料,本文教导并要求保护的改进循环吸热烃转化工艺可以用于涉及其中期望在催化剂床内进行温度控制的吸热反应的任何工艺。此类工艺包括但不限于固定床链烷烃脱氢、流化床链烷烃脱氢和流化床乙苯脱氢。在这些工艺中,催化剂和与产热材料组合的催化剂可以分层放置或均匀混合。类似地,可以预料,组合了产热第二组分的反应特异性催化剂的组合可以用于其中期望在催化剂床内进行温度控制的任何工艺。应理解,催化剂组合物和特定处理条件可以发生变化,而不会超出这一发明的范围。
改进的吸热烃转化工艺专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0